diff --git a/CITATIONS.md b/CITATIONS.md
index efaabc59..dd8a8e3a 100644
--- a/CITATIONS.md
+++ b/CITATIONS.md
@@ -13,12 +13,12 @@
* [PGS Catalog API](https://pubmed.ncbi.nlm.nih.gov/33692568/)
> Lambert SA, Gil L, Jupp S, Ritchie SC, Xu Y, Buniello A, McMahon A, Abraham G, Chapman M, Parkinson H, Danesh J. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nature Genetics. 2021 Apr;53(4):420-5. doi: 10.1038/s41588-021-00783-5. PubMed PMID: 33692568.
-* [PLINK 1](https://pubmed.ncbi.nlm.nih.gov/17701901/)
- > Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. The American journal of human genetics. 2007 Sep 1;81(3):559-75. doi: 10.1086/519795. PubMed PMID: 17701901. PubMed Central PMCID: PMC1950838.
-
* [PLINK 2](https://pubmed.ncbi.nlm.nih.gov/25722852/)
> Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015 Dec 1;4(1):s13742-015. doi: 10.1186/s13742-015-0047-8. PubMed PMID: 25722852. PubMed Central PMCID: PMC4342193.
+* [FRAPOSA](https://pubmed.ncbi.nlm.nih.gov/32196066/)
+ > Zhang, D., et al. (2020) Fast and robust ancestry prediction using principal component analysis. Bioinformatics 36(11):3439–3446. https://doi.org/10.1093/bioinformatics/btaa152
+
## Software packaging/containerisation tools
* [Anaconda](https://anaconda.com)
diff --git a/README.md b/README.md
index ac2e7a9e..b64462fe 100644
--- a/README.md
+++ b/README.md
@@ -5,9 +5,9 @@
[](https://doi.org/10.5281/zenodo.7577371)
[](https://www.nextflow.io/)
-[](https://docs.conda.io/en/latest/)
[](https://www.docker.com/)
[](https://sylabs.io/docs/)
+[](https://docs.conda.io/en/latest/)
## Introduction
@@ -19,9 +19,11 @@ and/or user-defined PGS/PRS.
## Pipeline summary
-  +
+ 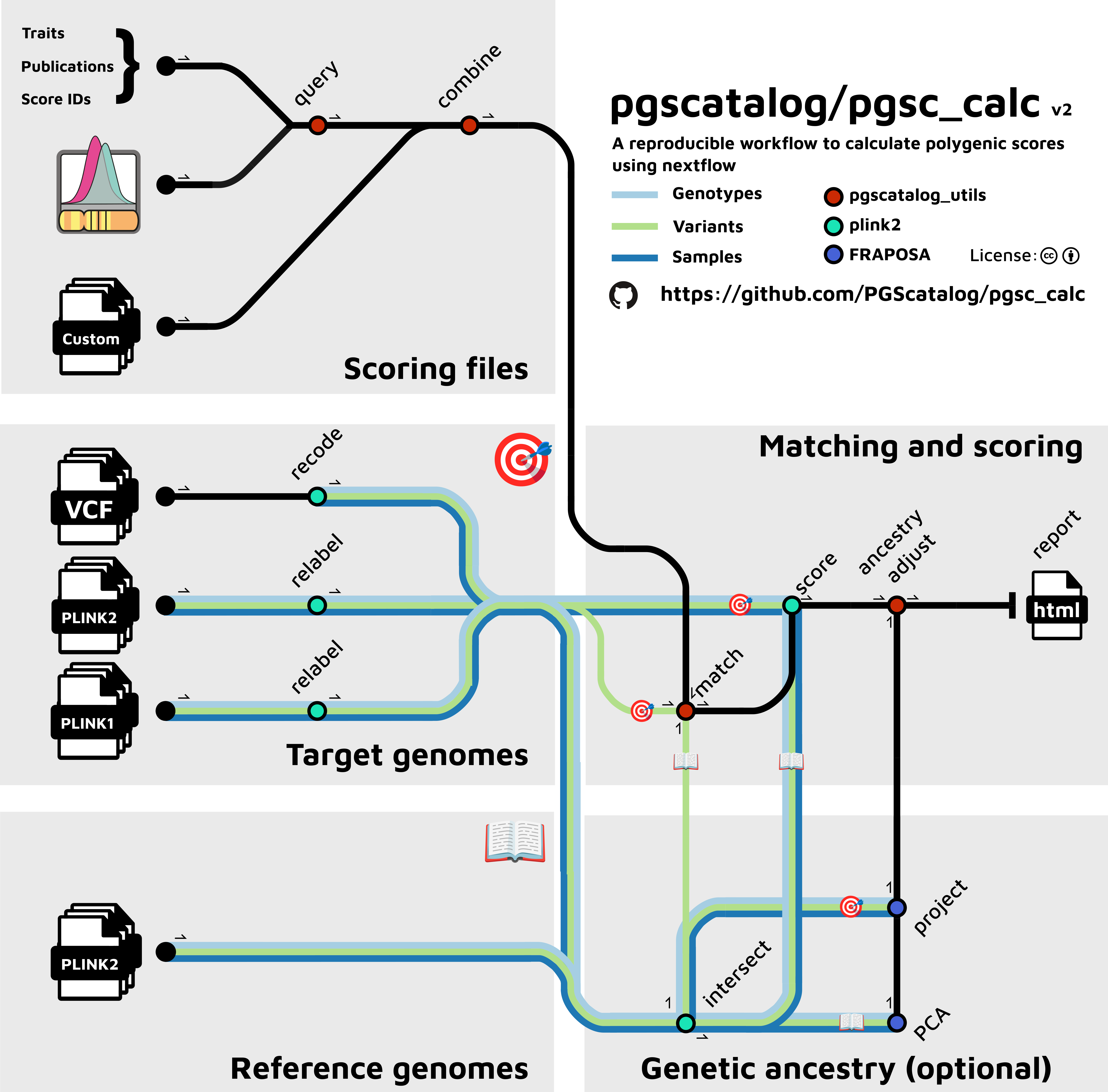
+The workflow performs the following steps:
+
* Downloading scoring files using the PGS Catalog API in a specified genome build (GRCh37 and GRCh38).
* Reading custom scoring files (and performing a liftover if genotyping data is in a different build).
* Automatically combines and creates scoring files for efficient parallel computation of multiple PGS
@@ -29,13 +31,14 @@ and/or user-defined PGS/PRS.
* Calculates PGS for all samples (linear sum of weights and dosages)
* Creates a summary report to visualize score distributions and pipeline metadata (variant matching QC)
-### Features in development
+And optionally:
+
+- Genetic Ancestry: calculate similarity of target samples to populations in a
+ reference dataset ([1000 Genomes (1000G)](http://www.nature.com/nature/journal/v526/n7571/full/nature15393.html)), using principal components analysis (PCA)
+- PGS Normalization: Using reference population data and/or PCA projections to report
+ individual-level PGS predictions (e.g. percentiles, z-scores) that account for genetic ancestry
-- *Genetic Ancestry*: calculate similarity of target samples to populations in a
- reference dataset (e.g. [1000 Genomes (1000G)](http://www.nature.com/nature/journal/v526/n7571/full/nature15393.html),
- [Human Genome Diversity Project (HGDP)](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7115999/)) using principal components analysis (PCA).
-- *PGS Normalization*: Using reference population data and/or PCA projections to report
- individual-level PGS predictions (e.g. percentiles, z-scores) that account for genetic ancestry.
+See documentation for a list of planned [features under development](https://pgsc-calc.readthedocs.io/en/latest/index.html#Features-under-development).
## Quick start
@@ -87,7 +90,7 @@ manuscript describing the tool is in preparation. In the meantime if you use the
tool we ask you to cite the repo and the paper describing the PGS Catalog
resource:
-- >PGS Catalog Calculator _(in development)_. PGS Catalog
+- >PGS Catalog Calculator _(preprint forthcoming)_. PGS Catalog
Team. [https://github.com/PGScatalog/pgsc_calc](https://github.com/PGScatalog/pgsc_calc)
- >Lambert _et al._ (2021) The Polygenic Score Catalog as an open database for
reproducibility and systematic evaluation. Nature Genetics. 53:420–425
diff --git a/docs/_templates/globaltoc.html b/docs/_templates/globaltoc.html
index 9a2d25c1..6fff7d2a 100644
--- a/docs/_templates/globaltoc.html
+++ b/docs/_templates/globaltoc.html
@@ -6,12 +6,12 @@ Contents
How-to guides
Reference guides
Explanations
Troubleshooting
@@ -32,6 +32,7 @@ Useful links
pgsc_calc Github
pgscatalog_utils Github
diff --git a/docs/changelog.rst b/docs/changelog.rst
index 27cf0ef4..9f9b42b9 100644
--- a/docs/changelog.rst
+++ b/docs/changelog.rst
@@ -8,7 +8,25 @@ will only occur in major versions with changes noted in this changelog.
.. _`semantic versioning`: https://semver.org/
-pgsc_calc v1.3.2 (2022-01-27)
+pgsc_calc v2.0.0 (2023-08-08)
+-----------------------------
+
+This major release features breaking changes to samplesheet structure to provide
+more flexible support for extra genomic file types in the future. Two major new
+features were implemented in this release:
+
+- Genetic ancestry group similarity is calculated to a population reference panel
+ (default: 1000 Genomes) when the ``--run_ancestry`` flag is supplied. This runs
+ using PCA and projection implemented in the ``fraposa_pgsc (v0.1.0)`` package.
+- Calculated PGS can be adjusted for genetic ancestry using empirical PGS distributions
+ from the most similar reference panel population or continuous PCA-based regressions.
+
+These new features are optional and don't run in the default workflow. Other features
+included in the release are:
+
+- Speed optimizations for PGS scoring (skipping allele frequency calculation)
+
+pgsc_calc v1.3.2 (2023-01-27)
-----------------------------
This patch fixes a bug that made some PGS Catalog scoring files incompatible
@@ -18,7 +36,7 @@ reporting the problem.
.. _`@j0n-a`: https://github.com/PGScatalog/pgsc_calc/issues/79
-pgsc_calc v1.3.1 (2022-01-24)
+pgsc_calc v1.3.1 (2023-01-24)
-----------------------------
This patch fixes a bug that breaks the workflow if all variants in one or more
diff --git a/docs/explanation/geneticancestry.rst b/docs/explanation/geneticancestry.rst
new file mode 100644
index 00000000..6a8be6a0
--- /dev/null
+++ b/docs/explanation/geneticancestry.rst
@@ -0,0 +1,171 @@
+.. _norm:
+
+Reporting and adjusting PGS in the context of genetic ancestry
+==============================================================
+
+v2 of the ``pgsc_calc`` pipeline introduces the ability to analyse the genetic ancestry of the individuals in your
+sampleset in comparison to a reference panel (default: 1000 Genomes) using principal component analysis (PCA). In this
+document we explain how the PCA is derived, and how it can be used to report polygenic scores that are adjusted or
+contextualized by genetic ancestry using multiple different methods.
+
+
+Motivation: PGS distributions and genetic ancestry
+--------------------------------------------------
+PGS are developed to measure an individual’s genetic predisposition to a disease or trait. A common way to express this
+is as a relative measure of predisposition (e.g. risk) compared to a reference population (often presented as a Z-score
+or percentile). The choice of reference population is important, as the mean and variance of a PGS can differ between
+different genetic ancestry groups (`Figure 1`_) as has been shown previously.\ [#Reisberg2017]_\ [#Martin2017]_
+
+.. _Figure 1:
+.. figure:: screenshots/p_SUM.png
+ :width: 450
+ :alt: Example PGS distributions stratified by population groups.
+
+ **Figure 1. Example of a PGS with shifted distributions in different ancestry groups.** Shown
+ here is the distribution of PGS000018 (metaGRS_CAD) calculated using the SUM method
+ in the 1000 Genomes reference panel, stratified by genetic ancestry groups (superpopulation labels).
+
+It is important to note that differences in the means between different ancestry groups do not necessarily correspond
+to differences in the risk (e.g., changes in disease prevalence, or mean biomarker values) between the populations.
+Instead, these differences are caused by changes in allele frequencies and linkage disequilibrium (LD) patterns between
+ancestry groups. This illustrates that genetic ancestry is important for determining relative risk, and multiple
+methods to adjust for these differences have been implemented within the pgsc_calc pipeline.
+
+Methods for reporting and adjusting PGS in the context of ancestry
+------------------------------------------------------------------
+.. _adjustment methods:
+
+When a PGS is being applied to a genetically homogenous population (e.g. cluster of individuals of similar genetic
+ancestry), then the standard method is to normalize the calculated PGS using the sample mean and standard
+deviation. This can be performed by running pgsc_calc and taking the Z-score of the PGS SUM. However, if you wish to
+adjust the PGS in order to remove the effects of genetic ancestry on score distributions than the
+``--run_ancestry`` method can combine your PGS with a reference panel of individuals (default 1000 Genomes) and apply
+standard methods for adjusting PGS (`Figure 2`_). These methods both start by creating a PCA of the reference panel,
+and projecting individual(s) into the genetic ancestry space to determine their placement and most similar population.
+The two groups of methods (empirical and continuous PCA-based) use these data and the calculated PGS to report the PGS.
+
+.. _Figure 2:
+.. figure:: screenshots/Fig_AncestryMethods.png
+ :width: 1800
+ :alt: Schematic figure detailing methods for contextualizing or adjusting PGS in the context of genetic ancestry.
+
+ **Figure 2. Schematic figure detailing empirical and PCA-based methods for contextualizing or adjusting PGS
+ with genetic ancestry.** Data is for the normalization of PGS000018 (metaGRS_CAD) in 1000 Genomes,
+ when applying ``pgsc_calc --run_ancestry`` to data from the Human Genome Diversity Project (HGDP) data.
+
+
+Empirical methods
+~~~~~~~~~~~~~~~~~
+A common way to report the relative PGS for an individual is by comparing their score with a distribution
+of scores from genetically similar individuals (similar to taking a Z-score within a genetically homogenous population
+above).\ [#ImputeMe]_ To define the correct reference distribution of PGS for an individual we first train a classifier
+to predict the population labels (pre-defined ancestry groups from the reference panel) using PCA loadings in the
+reference panel. This classifier is then applied to individuals in the target dataset to identify the population they are
+most similar to in genetic ancestry space. The relative PGS for each individual is calculated by comparing the
+calculated PGS to the distribution of PGS in the most similar population in the reference panel and reporting it as a
+percentile (output column: ``percentile_MostSimilarPop``) or as a Z-score (output column: ``Z_MostSimilarPop``).
+
+
+PCA-based methods
+~~~~~~~~~~~~~~~~~
+Another way to remove the effects of genetic ancestry on PGS distributions is to treat ancestry as a continuum
+(represented by loadings in PCA-space) and use regression to adjust for shifts therein. Using regression has the
+benefit of not assigning individuals to specific ancestry groups, which may be particularly problematic for empirical
+methods when an individual has an ancestry that is not represented within the reference panel. This method was first
+proposed by Khera et al. (2019)\ [#Khera2019]_ and uses the PCA loadings to adjust for differences in the means of PGS
+distributions across ancestries by fitting a regression of PGS values based on PCA-loadings of individuals of the
+reference panel. To calculate the normalized PGS the predicted PGS based on the PCA-loadings is subtracted from the PGS
+and normalized by the standard deviation in the reference population to achieve PGS distributions that are centred
+at 0 for each genetic ancestry group (output column: ``Z_norm1``), while not relying on any population labels during
+model fitting.
+
+The first method (``Z_norm1``) has the result of normalizing the first moment of the PGS distribution (mean); however,
+the second moment of the PGS distribution (variance) can also differ between ancestry groups. A second regression of
+the PCA-loadings on the squared residuals (difference of the PGS and the predicted PGS) can be fitted to estimate a
+predicted standard deviation based on genetic ancestry, as was proposed by Khan et al. (2022)\ [#Khan2022]_ and
+implemented within the eMERGE GIRA.\ [#GIRA]_ The predicted standard deviation (distance from the mean PGS based on
+ancestry) is used to normalize the residual PGS and get a new estimate of relative risk (output column: ``Z_norm2``)
+where the variance of the PGS distribution is more equal across ancestry groups and approximately 1.
+
+
+Implementation of ancestry steps within ``pgsc_calc``
+-----------------------------------------------------
+The ancestry methods are implemented within the ``--run_ancestry`` method of the pipeline (see :ref:`ancestry` for a
+how-to guide), and has the following steps:
+
+1. **Reference panel**: preparing and/or extracting data of the reference panel for use in the pipeline (see
+ :ref:`database` for details about downloading the existing reference [1000 Genomes] or setting up your own).
+
+2. **Variant overlap**: Identifying variants that are present in both the target genotypes and the reference panel.
+ Uses the ``INTERSECT_VARIANTS`` module.
+
+3. **PGS Calculation**:
+ 1. **Preparing scoring files**: in order to normalize the PGS the score has to be calculated on identical variant sets both datasets.
+ The list of overlapping variants between the reference and target datasets are supplied to the ``MATCH_COMBINE``
+ module to exclude scoring file variants that are matched only in the target genotypes.
+
+ 2. **PGS Scoring**: those scoring files are then supplied to the ``PLINK2_SCORE`` module, along with allele
+ frequency information from the reference panel to ensure consistent scoring of the PGS SUMs across datasets.
+ The scoring is made efficient by scoring all PGS in parallel.
+
+4. **PCA of the reference panel**
+ 1. **Preparing reference panel for PCA**: the reference panel is filtered to unrelated samples with standard filters
+ for variant-level QC (SNPs in Hardy–Weinberg equilibrium [p > 1e-04] that are bi-allelic and non-ambiguous,
+ with low missingness [<10%], and minor allele frequency [MAF > 5%]) and sample-quality (missingness <10%).
+ LD-pruning is then applied to the variants and sample passing these checks (r\ :sup:`2` threshold = 0.05), excluding
+ complex regions with high LD (e.g. MHC). These methods are implemented in the ``FILTER_VARIANTS`` module.
+
+ 2. **PCA**: the LD-pruned variants of the unrelated samples passing QC are then used to define the PCA space of the
+ reference panel (default: 10 PCs) using `FRAPOSA`_ (Fast and Robust Ancestry Prediction by using Online singular
+ value decomposition and Shrinkage Adjustment).\ [#zhangfraposa]_ This is implemented in the ``FRAPOSA_PCA``
+ module. *Note: it is important to inspect the PCA in the report to make sure that it looks correct and places
+ the different reference populations correctly.*
+
+5. **Projecting target samples into the reference PCA space**: the PCA of the reference panel (variant-PC loadings, and
+ reference sample projections) are then used to determine the placement of the target samples in the PCA space using
+ projection. Naive projection (using loadings) is prone to shrinkage which biases the projection of individuals
+ towards the null of an existing space, which would introduce errors into PCA-loading based adjustments of PGS.
+ For less biased projection of individuals into the reference panel PCA space we use the **online augmentation,
+ decomposition and Procrustes (OADP)** method of the `FRAPOSA`_ package.\ [#zhangfraposa]_ We chose to implement
+ PCA-based projection over derivation of the PCA space on a merged target and reference dataset to ensure that the
+ composition of the target doesn't impact the structure of the PCA. This is implemented in the ``FRAPOSA_OADP`` module.
+ *Note: the quality of the projection should be visually assessed in the report - we plan to furhter optimize the QC
+ in this step in future versions.*
+
+6. **Ancestry analysis**: the calculated PGS (SUM), reference panel PCA, and target sample projection into the PCA space
+ are supplied to a script that performs the analyses needed to adjust the PGS for genetic ancestry. This
+ functionality is implemented within the ``ANCESTRY_ANALYSIS`` module and tool of our `pgscatalog_utils`_ package,
+ and includes:
+
+ 1. **Genetic similarity analysis**: first each individual in the target sample is compared to the reference panel to
+ determine the population that they are most genetically similar to. By default this is done by fitting a
+ RandomForest classifier to predict reference panel population assignments using the PCA-loadings (default:
+ 10 PCs) and then applying the classifier to the target samples to identify the most genetically similar
+ population in the reference panel (e.g. highest-probability). Alternatively, the Mahalanobis distance between
+ each individual and each reference population can be calculated and used to identify the most similar reference
+ population (minimum distance). The probability of membership for each reference population and most similar
+ population assignments are recorded and output for all methods.
+
+ 2. **PGS adjustment**: the results of the genetic similarity analysis are combined with the PCA-loadings and
+ calculated PGS to perform the `adjustment methods`_ described in the previous section. To perform the
+ **empirical adjusments** (``percentile_MostSimilarPop``, ``Z_MostSimilarPop``) the PGS and the population
+ labels are used. To perform the **PCA-based adjusments** (``Z_norm1``, ``Z_norm2``) only the PGS and
+ PCA-loadings are used.
+
+7. **Reporting & Outputs**: the final results are output to txt files for further analysis, and an HTML report with
+ visualizations of the PCA data and PGS distributions (see :ref:`interpret` for additional details). It is important
+ to inspect the PCA projections and post-adjustment distributions of PGS before using these data in any downstream
+ analysis.
+
+.. _`FRAPOSA`: https://github.com/PGScatalog/fraposa_pgsc
+.. _`pgscatalog_utils`: https://github.com/PGScatalog/pgscatalog_utils
+
+
+.. rubric:: Citations
+.. [#Reisberg2017] Reisberg S., et al. (2017) Comparing distributions of polygenic risk scores of type 2 diabetes and coronary heart disease within different populations. PLoS ONE 12(7):e0179238. https://doi.org/10.1371/journal.pone.0179238
+.. [#Martin2017] Martin, A.R., et al. (2017) Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. The American Journal of Human Genetics 100(4):635-649. https://doi.org/10.1016/j.ajhg.2017.03.004
+.. [#ImputeMe] Folkersen, L., et al. (2020) Impute.me: An Open-Source, Non-profit Tool for Using Data From Direct-to-Consumer Genetic Testing to Calculate and Interpret Polygenic Risk Scores. Frontiers in Genetics 11:578. https://doi.org/10.3389/fgene.2020.00578
+.. [#Khera2019] Khera A.V., et al. (2019) Whole-Genome Sequencing to Characterize Monogenic and Polygenic Contributions in Patients Hospitalized With Early-Onset Myocardial Infarction. Circulation 139:1593–1602. https://doi.org/10.1161/CIRCULATIONAHA.118.035658
+.. [#Khan2022] Khan, A., et al. (2022) Genome-wide polygenic score to predict chronic kidney disease across ancestries. Nature Medicine 28:1412–1420. https://doi.org/10.1038/s41591-022-01869-1
+.. [#GIRA] Linder, J.E., et al. (2023) Returning integrated genomic risk and clinical recommendations: The eMERGE study. Genetics in Medicine 25(4):100006. https://doi.org/10.1016/j.gim.2023.100006.
+.. [#zhangfraposa] Zhang, D., et al. (2020) Fast and robust ancestry prediction using principal component analysis. Bioinformatics 36(11):3439–3446. https://doi.org/10.1093/bioinformatics/btaa152
diff --git a/docs/explanation/index.rst b/docs/explanation/index.rst
index 2f319e4e..a89bd1eb 100644
--- a/docs/explanation/index.rst
+++ b/docs/explanation/index.rst
@@ -8,3 +8,4 @@ Explanation
output
normalisation
+ plink2
diff --git a/docs/explanation/normalisation.rst b/docs/explanation/normalisation.rst
deleted file mode 100644
index ff107bd6..00000000
--- a/docs/explanation/normalisation.rst
+++ /dev/null
@@ -1,6 +0,0 @@
-.. _norm:
-
-Normalisation
-=============
-
-
diff --git a/docs/explanation/output.rst b/docs/explanation/output.rst
index 159771c7..003c8d92 100644
--- a/docs/explanation/output.rst
+++ b/docs/explanation/output.rst
@@ -1,7 +1,7 @@
.. _interpret:
-``pgsc_calc`` Outputs & results
-===============================
+``pgsc_calc`` Outputs & report
+==============================
The pipeline outputs are written to a results directory (``--outdir`` default is
@@ -10,72 +10,103 @@ The pipeline outputs are written to a results directory (``--outdir`` default is
.. code-block:: console
results
- ├── match (scoring files and variant match metadata)
- ├── pipeline_info (nextflow pipeline execution data)
- └── score (calculated PGS with summary report)
+ ├── [sampleset] (directory with results for your data)
+ │ ├── match (scoring files and variant match metadata)
+ │ └── score (calculated PGS with summary report)
+ └── pipeline_info (nextflow pipeline execution data)
``score/``
----------
Calculated scores are stored in a gzipped-text space-delimted text file called
-``aggregated_scores.txt.gz``. Each row represents an individual, and there should
-be at least three columns with the following headers:
+``[sampleset]_pgs.txt.gz``. The data is presented in long form where each PGS for an individual is presented on a
+seperate row (``length = n_samples*n_pgs``), and there will be at least four columns with the following headers:
-- ``dataset``: the name of the input sampleset
-- ``IID``: the identifier of each sample within the dataset
-- ``[PGS NAME]_SUM``: reports the weighted sum of *effect_allele* dosages multiplied by their *effect_weight*
- for each matched variant in the scoring file. The column name will be different depending on the scores
- you have chosen to use (e.g. ``PGS000001_SUM``).
+- ``sampleset``: the name of the input sampleset, or ``reference`` for the panel.
+- ``IID``: the identifier of each sample within the dataset.
+- ``PGS``: the accession ID of the PGS being reported.
+- ``SUM``: reports the weighted sum of *effect_allele* dosages multiplied by their *effect_weight*
+ for each matched variant in the scoring file for the PGS.
-At least one score must be present in this file (the third column). Extra columns might be
-present if you calculated more than one score, or if you calculated the PGS on a dataset with a
-small sample size (n < 50, in this cases a column named ``[PGS NAME]_AVG`` will be added that
-normalizes the PGS using the number of non-missing genotypes to avoid using allele frequency data
-from the target sample).
+If you have run the pipeline **without** using ancestry information the following columns may be present:
+
+- ``DENOM``: the number of non-missing genotypes used to calculate the PGS for this individual.
+- ``AVG``: normalizes ``SUM`` by the ``DENOM`` field (displayed when you calculate the PGS on a small sample size n<50
+ to avoid using unreliable allele frequency estimates for missing genotypes in the target sample.
+
+If you have run the pipeline **using ancestry information** (``--run_ancesty``) the following columns may be present
+depending on the ancestry adjustments that were run (see `adjustment methods`_ for more details):
+
+- ``percentile_MostSimilarPop``: PGS reported as a percentile of the distribution for the Most Similar Population
+- ``Z_MostSimilarPop``: PGS reported as a Z-score in reference to the mean/sd of the Most Similar Population
+- ``Z_norm1``: PGS adjusted to have mean 0 across ancestry groups (result of regressing *PGS ~ PCs*)
+- ``Z_norm2``: PGS adjusted to have mean 0 and unit variance across ancestry groups (result of regressing
+ *resid(PGS)^2 ~ PCs*)
Report
~~~~~~
-A summary report is also available (``report.html``). The report should open in
-a web browser and contains useful information about the PGS that were applied,
-how well the variants match with the genotyping data, and some simple graphs
-displaying the distribution of scores in your dataset(s) as a density plot.
+A summary report is also provided for your samples (``report.html``). The report should open in a web browser and
+contains useful information about the PGS that were applied, how well the variants in your target dataset match with the
+reference panel and scoring files, a summary of the computed genetic ancestry data, and some simple graphs displaying
+the distribution of scores in your dataset(s) as a density plot. Some of the sections are only displayed with
+``--run_ancestry``, but we show them all here for reference.
-The fist section of the report reproduces the nextflow command, and metadata (imported
-from the PGS Catalog for each PGS ID) describing the scoring files that were applied
-to your sampleset(s):
+The fist section of the report reproduces the nextflow command, and scoring file metadata (imported from the PGS Catalog
+for each PGS ID) describing the scoring files that were applied to your sampleset(s):
-.. image:: screenshots/Report_1_Header.png
+.. figure:: screenshots/Report_1_Header.png
:width: 600
:alt: Example PGS Catalog Report: header sections
-Within the scoring file metadata section are two tables describing how well the variants within
-each scoring file match with target sampleset(s). The first table provides a summary of the
-number and percentage of variants within each score that have been matched, and whether that
-score passed the ``--min_overlap`` threshold (Passed Matching column) for calculation. The second
-table provides a more detailed summary of variant matches broken down by types of variants (strand ambiguous,
+ **Figure 1. Example of pgsc_calc header.**
+
+
+The next section reports how the variants in the target sampleset match the other data. The first table describes the
+number of variants in the target dataset that overlap with the reference panel (*only present with* ``--run_ancestry``).
+The second table provides a summary of the number and percentage of variants within each score that have been matched,
+and whether that score passed the ``--min_overlap`` threshold (Passed Matching column) for calculation. The third
+table provides a more detailed summary of variant matches broken down by types of variants (e.g., strand ambiguous,
multiallelic, duplicates) for the matched, excluded, and unmatched variants (see ``match/`` section for details):
-.. image:: screenshots/Report_2_VariantMatching.png
+.. figure:: screenshots/Report_2_VariantMatching.png
:width: 600
:alt: Example PGS Catalog Report: Variant matching/qc tables (summary & detailed)
-The final section shows an example of the results table that contains the sample identifiers and
-calculated PGS in the *Score extract* panel. A visual display of the PGS distribution for a set of example
-score(s) (up to 6) is provided in the *Density plot* panel which can be helpful for looking at the distributions in
-multiple dataset(s):
+ **Figure 2. Example of variant matching summaries in the pgsc_calc report.**
-.. image:: screenshots/Report_3_Scores.png
+
+The next section describes the results of the genetic ancestry analysis of the target genotypes with the reference
+panel data. It first displays a snippet of the ``[sampleset]_popsimilarity.txt.gz`` file for reference. A visual display
+of the projection of the target data into the reference panel PCA space is plot for the first 6 PCs, where the target
+samples are coloured according to the population that they are most similar to in the reference panel. A table
+describing the distribution of ancestries within the reference panel and proportions of the target samples who are most
+similar to those populations is also provided.
+
+.. figure:: screenshots/Report_3_PCA.png
:width: 600
- :alt: Example PGS Catalog Report: Variant matching/qc tables (summary & detailed)
+ :alt: Example PGS Catalog Report: PCA plot of genetic ancestry data
+
+ **Figure 3. Visualization of genetic ancestry analysis within the report.**
+
+
+The final section shows an example of the main results dataframe that contains the sample identifiers and
+calculated PGS in the *Score extract* section. A visual display of the PGS distribution for a set of example
+score(s) (up to 6) is provided in the *Density plot* panel which can be helpful for looking at the distributions of the
+scores in the target and reference dataset and how it changes for difference PGS adjustment methods:
+
+.. figure:: screenshots/Report_4_Scores.png
+ :width: 600
+ :alt: Example PGS Catalog Report: table and density plots of score distributions
+
+ **Figure 4. Example of the** ``[sampleset]_pgs.txt.gz`` **table and plots of PGS distributions.**
``match/``
----------
-This directory contains information about the matching of scoring file variants to
-the genotyping data (samplesets). First a summary file (also displayed in the report)
-details whether each scoring file passes the minimum variant matching threshold, and
-the types of variants that were included in the score:
+This directory contains information about the matching of scoring file variants to your genotyping data (sampleset).
+First a summary file (also displayed in the report) details whether each scoring file passes the minimum variant
+matching threshold, and the types of variants that were included in the score:
.. list-table:: ``[sampleset]_summary.csv`` metadata
:widths: 20, 20, 60
@@ -96,34 +127,43 @@ the types of variants that were included in the score:
and is included in the final scoring file.
* - Match type
- ``match_status``
- - Indicates whether the variant is matched (included in the final scoring file),
- excluded (matched but removed based on variant filters), or unmatched.
+ - Indicates whether the variants are matched (included in the final scoring file), excluded (matched but removed
+ based on variant filters), or unmatched.
* - Ambiguous
- ``ambiguous``
- - True/False flag indicating whether the matched variant is strand-ambiguous (e.g. A/T and C/G variants).
+ - True/False flag indicating whether the matched variants are strand-ambiguous (e.g. A/T and C/G variants).
* - Multiallelic
- ``is_multiallelic``
- - True/False flag indicating whether the matched variant is multi-allelic (multiple ALT alleles).
+ - True/False flag indicating whether the matched variants are multi-allelic (multiple ALT alleles).
* - Multiple potential matches
- ``duplicate_best_match``
- - True/False flag indicating whether a single scoring file variants has multiple potential matches to the target genome.
- This usually occurs when the variant has no other_allele, and with variants that have different REF alleles.
+ - True/False flag indicating whether a single scoring file variant has multiple potential matches to the target genome.
+ This usually occurs when the variant has no other/non-effect allele, and with variants that have different
+ REF alleles.
* - Duplicated matched variants
- ``duplicate_ID``
- True/False flag indicating whether multiple scoring file variants match a single target ID. This usually occurs
- when scoring files have been lifted across builds and two variants now point to the same position (e.g. rsID mergers).
+ when scoring files have been lifted across builds and two variants now point to the same position (e.g. rsID
+ mergers).
+ * - Matches strand flip
+ - ``match_flipped``
+ - True/False flag indicating whether the scoring file variant is originally reported on the opposite strand (and
+ thus flipped to match)
+ * - Variant in reference panel
+ - ``match_IDs``
+ - True/False flag indicating whether the variant from the scoring file that is matched in the target samples is
+ also present in the variants that overlap with the reference population panel (required for PGS adjustment).
* - n
- ``count``
- Number of variants with this combination of metadata (grouped by: ``[ match_status, ambiguous, is_multiallelic,
- duplicate_best_match, duplicate_ID]``
+ duplicate_best_match, duplicate_ID, match_flipped, match_IDs]``
* - %
- ``percent``
- Percent of the scoring file's variants that have the combination of metadata in count.
-The log file is a :term:`CSV` that contains all possible matches
-for each variant in the combined input scoring files. This information is useful to debug a
-score that is causing problems. Columns contain information about how each
+The log file is a :term:`CSV` that contains all possible matches for each variant in the combined input scoring files.
+This information is useful to debug a score that is causing problems. Columns contain information about how each
variant was matched against the target genomes:
@@ -170,11 +210,19 @@ variant was matched against the target genomes:
- True/False flag indicating whether the matched variant is multi-allelic (multiple ALT alleles).
* - ``ambiguous``
- True/False flag indicating whether the matched variant is strand-ambiguous (e.g. A/T and C/G variants).
+ * - ``match_flipped``
+ - True/False flag indicating whether the matched variant is on the opposite strand (flipped).
+ * - ``best_match``
+ - True/False flag indicating whether this the best ``match_type`` for the current scoring file variant.
+ * - ``exclude``
+ - True/False flag indicating whether this matched variant is excluded from the final scoring file.
* - ``duplicate_best_match``
- True/False flag indicating whether a single scoring file variants has multiple potential matches to the target genome.
This usually occurs when the variant has no other_allele, and with variants that have different REF alleles.
* - ``duplicate_ID``
- True/False flag indicating whether multiple scoring file variants match a single target ID.
+ * - ``match_IDs``
+ - True/False flag indicating whether the matched variant is also found in the reference panel genotypes.
* - ``match_status``
- Indicates whether the variant is *matched* (included in the final scoring file), *excluded* (matched but removed
based on variant filters), *not_best* (a different match candidate was selected for this scoring file variant),
@@ -183,12 +231,10 @@ variant was matched against the target genomes:
- Name of the sampleset/genotyping data.
-Processed scoring files are also present in this directory. Briefly, variants in
-the scoring files are matched against the target genomes. Common variants across
-different scores are combined (left joined, so each score is an additional
-column). The combined scores are then partially split to overcome PLINK2
-technical limitations (e.g. calculating different effect types such as dominant
-/ recessive). Once scores are calculated from these partially split scoring
+Processed scoring files are also present in this directory. Briefly, variants in the scoring files are matched against
+the target genomes. Common variants across different scores are combined (left joined, so each score is an additional
+column). The combined scores are then partially split to overcome PLINK2 technical limitations (e.g. calculating
+different effect types such as dominant/recessive). Once scores are calculated from these partially split scoring
files, scores are aggregated to produce the final results in ``score/``.
``pipeline_info/``
diff --git a/docs/explanation/plink2.rst b/docs/explanation/plink2.rst
new file mode 100644
index 00000000..033171f4
--- /dev/null
+++ b/docs/explanation/plink2.rst
@@ -0,0 +1,45 @@
+.. _plink2:
+
+Why not just use ``plink2 --score``?
+====================================
+
+You might be curious what ``pgsc_calc`` does that ``plink2 --score`` doesn't. We
+use ``plink2`` internally to calculate all scores but offer some extra features:
+
+- We match the variants from scoring files (often from different genome builds) against
+ the target genome using multiple strategies, taking into account the strand alignment,
+ ambiguous or multi-allelic matches, duplicated variants/matches, and overlaps between
+ datasets (e.g. with a reference for ancestry).
+
+- One important output is an auditable log that covers the union of all variants
+ across the scoring files, tracking how the variants matched the target genomes and
+ provide reasons why they are excluded from contributing to the final calculated scores
+ based on user-specified settings and thresholds for variant matching.
+
+- From the matched variants the workflow outputs a new set of scoring files
+ which are used by plink2 for scoring. The new scoring files combine multiple PGS in
+ a single file to calculate scores in parallel. These scoring files are automatically
+ split by effect type and across duplicate variant IDs with different effect alleles,
+ then the split scores are aggregated.
+
+- The pipeline also calculates genetic ancestry using a reference panel (default 1000 Genomes)
+ handling the data handling, variant matching, derivation of the PCA space, and projection of
+ target samples into the PCA space using robust methods (implemented in fraposa_pgsc_).
+
+- Scores can be adjusted using genetic ancestry data using multiple methods (see :ref:`norm`).
+
+.. _fraposa_pgsc: https://github.com/PGScatalog/fraposa_pgsc
+
+Summary
+-------
+
+- For a seasoned bioinformatician the workflow offers convenient integration with the PGS Catalog
+ and simplifies large scale PGS calculation on HPCs.
+
+- Genetic ancestry similarity calculations and adjustment of PGS using established methods
+ using reproducible code and datasets.
+
+- For a data scientist or somebody less familiar with the intricacies of PGS
+ calculation, ``pgsc_calc`` automates many of the complex steps and analysis choices needed to
+ calculate PGS using multiple software tools.
+
diff --git a/docs/explanation/screenshots/Fig_AncestryMethods.png b/docs/explanation/screenshots/Fig_AncestryMethods.png
new file mode 100644
index 00000000..5ea8381f
Binary files /dev/null and b/docs/explanation/screenshots/Fig_AncestryMethods.png differ
diff --git a/docs/explanation/screenshots/Report_1_Header.png b/docs/explanation/screenshots/Report_1_Header.png
index 63bb2bdd..7346957f 100644
Binary files a/docs/explanation/screenshots/Report_1_Header.png and b/docs/explanation/screenshots/Report_1_Header.png differ
diff --git a/docs/explanation/screenshots/Report_2_VariantMatching.png b/docs/explanation/screenshots/Report_2_VariantMatching.png
index 2e24a5ca..c7d8f13e 100644
Binary files a/docs/explanation/screenshots/Report_2_VariantMatching.png and b/docs/explanation/screenshots/Report_2_VariantMatching.png differ
diff --git a/docs/explanation/screenshots/Report_3_PCA.png b/docs/explanation/screenshots/Report_3_PCA.png
new file mode 100644
index 00000000..4efc6351
Binary files /dev/null and b/docs/explanation/screenshots/Report_3_PCA.png differ
diff --git a/docs/explanation/screenshots/Report_3_Scores.png b/docs/explanation/screenshots/Report_3_Scores.png
deleted file mode 100644
index d58ca487..00000000
Binary files a/docs/explanation/screenshots/Report_3_Scores.png and /dev/null differ
diff --git a/docs/explanation/screenshots/Report_4_Scores.png b/docs/explanation/screenshots/Report_4_Scores.png
new file mode 100644
index 00000000..90674119
Binary files /dev/null and b/docs/explanation/screenshots/Report_4_Scores.png differ
diff --git a/docs/explanation/screenshots/p_SUM.png b/docs/explanation/screenshots/p_SUM.png
new file mode 100644
index 00000000..b501ae51
Binary files /dev/null and b/docs/explanation/screenshots/p_SUM.png differ
diff --git a/docs/getting-started.rst b/docs/getting-started.rst
index 4c891456..c7293752 100644
--- a/docs/getting-started.rst
+++ b/docs/getting-started.rst
@@ -5,10 +5,18 @@
Getting started
===============
-``pgsc_calc`` requires Nextflow and one of Docker, Singularity, or
-Anaconda. You will need a POSIX compatible system, like Linux or macOS, to run ``pgsc_calc``.
+``pgsc_calc`` has a few important software dependencies:
-1. Start by `installing nextflow`_:
+* Nextflow
+* Docker, Singularity, or Anaconda
+* Linux or macOS
+
+Without these dependencies installed you won't be able to run ``pgsc_calc``.
+
+Step by step setup
+------------------
+
+1. `Install nextflow`_:
.. code-block:: console
@@ -19,59 +27,45 @@ Anaconda. You will need a POSIX compatible system, like Linux or macOS, to run `
$ mv nextflow ~/bin/
-2. Next, `install Docker`_, `Singularity`_, or `Anaconda`_
-
-3. Finally, check Nextflow is working:
+2. Next, `install Docker`_, `Singularity`_, or `Anaconda`_ (Docker or
+ Singularity are best)
-.. code-block:: console
-
- $ nextflow run pgscatalog/pgsc_calc --help
-
-And check if Docker, Singularity, or Anaconda are working by running the
-workflow with bundled test data and replacing ```` in
-the command below with the specific container manager you intend to use:
+3. Run the ``pgsc_calc`` test profile:
.. code-block:: console
- $ nextflow run pgscatalog/pgsc_calc -profile test,
+ $ nextflow run pgscatalog/pgsc_calc -profile test,
-.. _`installing nextflow`: https://www.nextflow.io/docs/latest/getstarted.html
+.. _`Install nextflow`: https://www.nextflow.io/docs/latest/getstarted.html
.. _`install Docker`: https://docs.docker.com/engine/install/
.. _`Singularity`: https://sylabs.io/guides/3.0/user-guide/installation.html
.. _`Anaconda`: https://docs.conda.io/projects/conda/en/latest/user-guide/install/index.html
-.. note:: Replace ```` with what you have installed on
- your computer (e.g., ``docker``, ``singularity``, or ``conda``). These
- options are mutually exclusive!
+.. note:: Remember to replace ```` one of ``docker``, ``singularity``, or ``conda``
-4. (Optional) Download the reference database from the PGS Catalog FTP:
+.. warning:: If you have an ARM processor (like Apple sillicon) please check :ref:`arm`
-.. code-block:: console
- $ wget https://ftp.pgscatalog.org/path/to/reference.tar.zst
-
-.. warning::
- - The reference database is required to run ancestry similarity analysis
- and to normalise calculated PGS
- - This getting started guide assumes you've downloaded and want to run the
- ancestry components of the workflow
- - If you don't want to run ancestry analysis, don't include the ``--ref``
- parameter from the examples below. Instead, add the ``--skip_ancestry``
- parameter.
+Please note the test profile genomes are not biologically meaningful, won't
+produce valid scores, and aren't compatible with scores on the PGS Catalog. We
+provide these genomes to make checking installation and automated testing
+easier.
Calculate your first polygenic scores
-=====================================
+-------------------------------------
-If you've completed the installation process that means you've already
-calculated some polygenic scores |:heart_eyes:| However, these scores were
-calculated using synthetic data from a single chromosome. Let's try calculating scores
-with your genomic data, which are probably genotypes from real people!
+If you've completed the setup guide successfully then you're ready to calculate
+scores with your genomic data, which are probably genotypes from real
+people. Exciting!
+.. warning:: The format of samplesheets changed in v2.0.0 to better accommodate
+ extra file formats in the future
+
.. warning:: You might need to prepare input genomic data before calculating
polygenic scores, see :ref:`prepare`
1. Set up samplesheet
----------------------
+~~~~~~~~~~~~~~~~~~~~~
First, you need to describe the structure of your genomic data in a standardised
way. To do this, set up a spreadsheet that looks like:
@@ -92,7 +86,7 @@ See :ref:`setup samplesheet` for more details.
2. Select scoring files
------------------------
+~~~~~~~~~~~~~~~~~~~~~~~
pgsc_calc makes it simple to work with polygenic scores that have been published
in the PGS Catalog. You can specify one or more scores using the ``--pgs_id``
@@ -123,16 +117,28 @@ then the pipeline will download a `harmonized (remapped rsIDs and/or lifted posi
scoring file(s) in the user-specified build of the genotyping datasets.
Custom scoring files can be lifted between genome builds using the ``--liftover`` flag, (see :ref:`liftover`
-for more information). An example would look like:
+for more information). If your custom PGS was in GRCh37 an example would look like:
.. code-block:: console
- ---scorefile MyPGSFile.txt --target_build GRCh38
+ ---scorefile MyPGSFile.txt --target_build GRCh38 --liftover
.. _harmonized (remapped rsIDs and/or lifted positions): https://www.pgscatalog.org/downloads/#dl_ftp_scoring_hm_pos
+3. (Optional) Download reference database
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
+
+To enable genetic ancestry similarity calculations and PGS normalisation,
+download our pre-built reference database:
+
+.. code-block:: console
+
+ $ wget https://ftp.ebi.ac.uk/pub/databases/spot/pgs/resources/pgsc_calc.tar.zst
+
+See :ref:`ancestry` for more details.
+
3. Putting it all together
---------------------------
+~~~~~~~~~~~~~~~~~~~~~~~~~~
For this example, we'll assume that the input genomes are in build GRCh37 and that
they match the scoring file genome build.
@@ -143,11 +149,13 @@ they match the scoring file genome build.
-profile \
--input samplesheet.csv --target_build GRCh37 \
--pgs_id PGS001229 \
- --ref pgsc_calc.tar.zst
+ --run_ancestry pgsc_calc.tar.zst
Congratulations, you've now (`hopefully`) calculated some scores!
|:partying_face:|
+.. tip:: Don't include ``--run_ancestry`` if you didn't download the reference database
+
After the workflow executes successfully, the calculated scores and a summary
report should be available in the ``results/score/`` directory in your current
working directory (``$PWD``) by default. If you're interested in more
@@ -157,8 +165,9 @@ If the workflow didn't execute successfully, have a look at the
:ref:`troubleshoot` section. Remember to replace ````
with the software you have installed on your computer.
+
4. Next steps & advanced usage
-------------------------------
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
The pipeline distributes with settings that easily allow for it to be run on a
personal computer on smaller datasets (e.g. 1000 Genomes, HGDP). The minimum
diff --git a/docs/how-to/ancestry.rst b/docs/how-to/ancestry.rst
new file mode 100644
index 00000000..18f1239d
--- /dev/null
+++ b/docs/how-to/ancestry.rst
@@ -0,0 +1,39 @@
+.. _ancestry:
+
+How do I normalise calculated scores across different genetic ancestry groups?
+==============================================================================
+
+Download reference data
+-----------------------
+
+The fastest method of getting started is to download our reference panel:
+
+.. code-block:: console
+
+ $ wget https://ftp.ebi.ac.uk/pub/databases/spot/pgs/resources/pgsc_calc.tar.zst
+
+The reference panel is based on 1000 Genomes. It was originally downloaded from
+the PLINK 2 `resources section`_. To minimise file size INFO annotations are
+excluded. KING pedigree corrections were enabled.
+
+.. _`resources section`: https://www.cog-genomics.org/plink/2.0/resources
+
+Bootstrap reference data
+~~~~~~~~~~~~~~~~~~~~~~~~
+
+It's possible to bootstrap (create from scratch) the reference data from the
+PLINK 2 data, which is how we create the reference panel tar. See
+:ref:`database`
+
+Genetic similarity analysis and score normalisation
+----------------------------------------------------------
+
+To enable genetic similarity analysis and score normalisation, just include the
+``--run_ancestry`` parameter when running the workflow:
+
+.. code-block:: console
+
+ $ nextflow run pgscatalog/pgsc_calc -profile test,docker \
+ --run_ancestry path/to/reference/pgsc_calc.tar.zst
+
+The ``--run_ancestry`` parameter requires the path to the reference database.
diff --git a/docs/how-to/bigjob.rst b/docs/how-to/bigjob.rst
index 3314e4d0..978bc419 100644
--- a/docs/how-to/bigjob.rst
+++ b/docs/how-to/bigjob.rst
@@ -11,6 +11,14 @@ these types of systems by creating and editing `nextflow .config files`_.
.. _nextflow .config files: https://www.nextflow.io/docs/latest/config.html
+.. warning:: ``--max_cpus`` and ``--max_memory`` don't increase the amount of
+ resources for each process. These parameters **cap the maximum
+ amount of resources** `a process can use`_. You need to edit
+ configuration files to increase process resources, as described
+ below.
+
+.. _`a process can use`: https://github.com/PGScatalog/pgsc_calc/issues/71#issuecomment-1423846928
+
Configuring ``pgsc_calc`` to use more resources locally
-------------------------------------------------------
@@ -123,9 +131,7 @@ instead:
.. note:: The name of the nextflow and singularity modules will be different in
your local environment
-.. note:: Think about enabling fast variant matching with ``--fast_match``!
-
-.. warning:: Make sure to copy input data to fast storage, and run the pipeline
+ .. warning:: Make sure to copy input data to fast storage, and run the pipeline
on the same fast storage area. You might include these steps in your
bash script. Ask your sysadmin for help if you're not sure what this
means.
diff --git a/docs/how-to/database.rst b/docs/how-to/database.rst
index dba6a957..d8f34b32 100644
--- a/docs/how-to/database.rst
+++ b/docs/how-to/database.rst
@@ -16,19 +16,19 @@ Download reference database
A reference database is available to download here:
-``ftp.pgscatalog.org/...``
+``https://ftp.ebi.ac.uk/pub/databases/spot/pgs/resources/pgsc_calc.tar.zst``
The database is about 7GB and supports both GRCh37 and GRCh38 input target
genomes.
-Once the reference database is included, remember you must include the ``--ref``
+Once the reference database is included, remember you must include the ``--run_ancestry``
parameter, which is a path to the reference database (see
:ref:`schema`).
(Optional) Create reference database
------------------------------------
-.. danger::
+.. Warning::
- Making a reference database from scratch can be slow and frustrating
- It's easiest to download the published reference database from the PGS Catalog FTP
diff --git a/docs/how-to/index.rst b/docs/how-to/index.rst
index 891aeb40..7e34b724 100644
--- a/docs/how-to/index.rst
+++ b/docs/how-to/index.rst
@@ -3,14 +3,6 @@
How-to guides
=============
-Reference database
-------------------
-
-.. toctree::
- :maxdepth: 1
-
- database
-
Calculating polygenic scores
----------------------------
@@ -21,6 +13,7 @@ Calculating polygenic scores
samplesheet
calculate_pgscatalog
multiple
+ ancestry
Making genomic data and scorefiles compatible
---------------------------------------------
@@ -32,15 +25,23 @@ Making genomic data and scorefiles compatible
liftover
effecttype
+Reference database
+------------------
+.. toctree::
+ :maxdepth: 1
+
+ database
+
Working in different environments
-----------------------------------
+---------------------------------
.. toctree::
:maxdepth: 1
bigjob
arm
+ offline
Working with complex datasets
-----------------------------
diff --git a/docs/how-to/offline.rst b/docs/how-to/offline.rst
new file mode 100644
index 00000000..3fb78e25
--- /dev/null
+++ b/docs/how-to/offline.rst
@@ -0,0 +1,116 @@
+.. _offline:
+
+How do I run pgsc_calc in an offline environment?
+=================================================
+
+pgsc_calc has been deployed on secure platforms like Trusted Research
+Environments (TREs). Running pgsc_calc is a little bit more difficult in this
+case. The basic set up approach is to:
+
+1. Download containers
+2. Download reference data
+3. Download scoring files
+
+And transfer everything to your offline environment.
+
+This guide assumes you've set up pgsc_calc and tested it in an online
+environment first.
+
+Every computing environment has different quirks and it can be difficult to get
+everything working correctly. Please feel free to `open a discussion on Github`_
+if you are having problems and we'll try our best to help you.
+
+.. _open a discussion on Github: https://github.com/PGScatalog/pgsc_calc/discussions
+
+Preload container images
+------------------------
+
+Docker
+~~~~~~
+
+Pull and save the docker images to local tar files in an online environment:
+
+.. code-block:: bash
+
+ $ cd /path/to/pgsc_calc
+ $ git grep 'ext.docker*' conf/modules.config | cut -f 2 -d '=' | xargs -L 2 echo | tr -d ' ' > images.txt
+ $ cat images.txt | xargs -I {} sh -c 'docker pull --platform linux/amd64 "$1"' - {}
+ $ mkdir -p docker/
+ $ cat images.txt | xargs -I {} sh -c 'docker save "$1" > docker/$(basename "$1").tar' - {}
+
+Tar files will have been saved to the ``docker/`` directory. Transfer this
+directory and load the container tars in the offline environment:
+
+.. code-block:: bash
+
+ $ find docker -name '*.tar'
+ $ find docker/ -name '*.tar' -exec sh -c 'docker load < {}' \;
+
+Singularity
+~~~~~~~~~~~
+
+Set ``NXF_SINGULARITY_CACHEDIR`` to the directory you want containers to
+download to:
+
+.. code-block:: bash
+
+ $ cd /path/to/pgsc_calc
+ $ export NXF_SINGULARITY_CACHEDIR=path/to/containers
+
+Then pull the images to the directory:
+
+.. code-block:: bash
+
+ $ mkdir -p $NXF_SINGULARITY_CACHEDIR
+ $ git grep 'ext.singularity*' conf/modules.config | cut -f 2 -d '=' | xargs -L 2 echo | tr -d ' ' > singularity_images.txt
+ $ cat singularity_images.txt | sed 's/oras:\/\///;s/https:\/\///;s/\//-/g;s/$/.img/;s/:/-/' > singularity_image_paths.txt
+ $ paste -d '\n'singularity_image_paths.txt singularity_images.txt | xargs -L 2 sh -c 'singularity pull --disable-cache --dir $NXF_SINGULARITY_CACHEDIR $0 $1'
+
+And transfer the directory to your offline environment.
+
+.. warning:: Remember to set ``NXF_SINGULARITY_CACHEDIR`` to the directory that
+ contains the downloaded containers on your offline system whenever
+ you run pgsc_calc, e.g.:
+
+ .. code-block:: bash
+
+ $ export NXF_SINGULARITY_CACHEDIR=path/to/containers
+ $ nextflow run main.nf -profile singularity ...
+
+Download reference data
+-----------------------
+
+Some small reference data is needed to run the calculator:
+
+* --hg19_chain https://hgdownload.cse.ucsc.edu/goldenpath/hg19/liftOver/hg19ToHg38.over.chain.gz
+* --hg38_chain https://hgdownload.soe.ucsc.edu/goldenPath/hg38/liftOver/hg38ToHg19.over.chain.gz
+
+To do ancestry-based score normalisation you'll need to download the reference
+panel too. See :ref:`norm`.
+
+Download scoring files
+----------------------
+
+It's best to manually download scoring files from the PGS Catalog in the correct
+genome build. Using PGS001229 as an example:
+
+https://ftp.ebi.ac.uk/pub/databases/spot/pgs/scores/PGS001229/ScoringFiles/
+
+.. code-block:: bash
+
+ $ PGS001229/ScoringFiles
+ ├── Harmonized
+ │ ├── PGS001229_hmPOS_GRCh37.txt.gz <-- the file you want
+ │ ├── PGS001229_hmPOS_GRCh37.txt.gz.md5
+ │ ├── PGS001229_hmPOS_GRCh38.txt.gz <-- or perhaps this one!
+ │ └── PGS001229_hmPOS_GRCh38.txt.gz.md5
+ ├── PGS001229.txt.gz
+ ├── PGS001229.txt.gz.md5
+ └── archived_versions
+
+These files can be transferred to the offline environment and provided to the
+workflow using the ``--scorefile`` parameter.
+
+.. tip:: If you're using multiple scoring files you must use quotes
+ e.g. ``--scorefile "path/to/scorefiles/PGS*.txt.gz"``
+
diff --git a/docs/how-to/prepare.rst b/docs/how-to/prepare.rst
index 84a14509..eb2fe4eb 100644
--- a/docs/how-to/prepare.rst
+++ b/docs/how-to/prepare.rst
@@ -7,10 +7,9 @@ Target genome data requirements
-------------------------------
.. note:: This workflow will work best with the output of an imputation server
- like `Michigan`_ or `TopMed`_
+ like `Michigan`_ or `TopMed`_.
-.. danger:: If you'd like to input WGS genomes, some extra preprocessing steps
- are required
+ If you'd like to input WGS genomes, some extra preprocessing steps are required.
.. _`Michigan`: https://imputationserver.sph.umich.edu/index.html
.. _`TopMed`: https://imputation.biodatacatalyst.nhlbi.nih.gov/
diff --git a/docs/how-to/samplesheet.rst b/docs/how-to/samplesheet.rst
index dc1f85d2..05b9a77f 100644
--- a/docs/how-to/samplesheet.rst
+++ b/docs/how-to/samplesheet.rst
@@ -7,6 +7,9 @@ A samplesheet describes the structure of your input genotyping datasets. It's ne
because the structure of input data can be very different across use cases (e.g.
different file formats, directories, and split vs. unsplit by chromosome).
+.. warning:: The format of samplesheets changed in v2.0.0 to better accommodate
+ additional file formats in the future
+
Samplesheet
-----------
@@ -67,6 +70,10 @@ Notes
genome build, no liftover/re-mapping of the genotyping data is performed
within the pipeline.
+.. note:: Your samplesheet can only contain one sampleset name. If you want to
+ run multiple large cohorts (e.g. 1000G and HGDP) then run the workflow
+ separately or combine the files.
+
Setting genotype field
~~~~~~~~~~~~~~~~~~~~~~
diff --git a/docs/index.rst b/docs/index.rst
index 9289a418..a0c032af 100644
--- a/docs/index.rst
+++ b/docs/index.rst
@@ -17,27 +17,33 @@ normalisation methods are also supported.
Workflow summary
----------------
-.. image:: https://user-images.githubusercontent.com/11425618/195053396-a3eaf31c-b3d5-44ff-a36c-4ef6d7958668.png
+.. image:: https://user-images.githubusercontent.com/11425618/257213197-f766b28c-0f75-4344-abf3-3463946e36cc.png
:width: 600
+ :align: center
:alt: `pgsc_calc` workflow diagram
|
-The workflow does the following steps:
+The workflow performs the following steps:
- Downloading scoring files using the PGS Catalog API in a specified genome build (GRCh37 and GRCh38).
- Reading custom scoring files (and performing a liftover if genotyping data is in a different build).
- Automatically combines and creates scoring files for efficient parallel
- computation of multiple PGS
-- Matching variants in the scoring files against variants in the target dataset (in plink bfile/pfile or VCF format)
-- Calculates PGS for all samples (linear sum of weights and dosages)
-- Creates a summary report to visualize score distributions and pipeline metadata (variant matching QC)
+ computation of multiple PGS.
+- Matches variants in the scoring files against variants in the target dataset (in plink bfile/pfile or VCF format).
+- Calculates PGS for all samples (linear sum of weights and dosages).
+- Creates a summary report to visualize score distributions and pipeline metadata (variant matching QC).
-And optionally:
+And optionally has additional functionality to:
+
+- Use a reference panel to obtain genetic ancestry data using PCA, and define the most similar population in the
+ reference panel for each target sample.
+- Report PGS using methods to adjust for genetic ancestry.
+
+.. tip:: To enable these optional steps, see :ref:`ancestry`
+
+See `Features under development`_ section for information about planned updates.
-- Using reference genomes to automatically assign the genetic ancestry of target
- genomes
-- Normalising calculated PGS to account for genetic ancestry
The workflow relies on open source scientific software, including:
@@ -51,6 +57,7 @@ A full description of included software is described in :ref:`containers`.
.. _PGS Catalog Utilities: https://github.com/PGScatalog/pgscatalog_utils
.. _FRAPOSA: https://github.com/PGScatalog/fraposa_pgsc
+
Quick example
-------------
@@ -108,27 +115,25 @@ Documentation
- :doc:`Get started`: install pgsc_calc and calculate some polygenic scores quickly
- :doc:`How-to guides`: step-by-step guides, covering different use cases
- :doc:`Reference guides`: technical information about workflow configuration
-
+- :doc:`Explanations`: more detailed explanations about PGS calculation and results
Changelog
---------
The :doc:`Changelog page` describes fixes and enhancements for each version.
-Features Under Development
+Features under development
--------------------------
-In the future, the calculator will include new features for PGS interpretation:
-
-- *Genetic Ancestry*: calculate similarity of target samples to populations in a
- reference dataset (e.g. `1000 Genomes (1000G)`_, `Human Genome Diversity Project (HGDP)`_)
- using principal components analysis (PCA).
-- *PGS Normalization*: Using reference population data and/or PCA projections to report
- individual-level PGS predictions (e.g. percentiles, z-scores) that account for genetic ancestry.
+These are some of the fetures and improvements we're planning for the ``pgsc_calc``:
-.. _1000 Genomes (1000G): http://www.nature.com/nature/journal/v526/n7571/full/nature15393.html
-.. _Human Genome Diversity Project (HGDP): https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7115999/
+- Improved population reference panels (merged 1000 Genomes & Human Genome Diversity Project (HGDP) for use
+ within the pipeline
+- Further optimizations to the PCA & ancestry similarity analysis steps focused on improving automatic QC
+- Performance improvments to make ``pgsc_calc`` work with 1000s of scoring files in paralell (e.g. integration
+ with `OmicsPred`_)
+.. _OmicsPred: https://www.omicspred.org
Credits
-------
@@ -148,14 +153,17 @@ are written by Benjamin Wingfield, Samuel Lambert, Laurent Gil with additional i
from Aoife McMahon (EBI). Development of new features, testing, and code review
is ongoing including Inouye lab members (Rodrigo Canovas, Scott Ritchie) and others. A
manuscript describing the tool is in preparation (see `Citations `_) and we
-welcome ongoing community feedback before then.
+welcome ongoing community feedback before then via our `discussion board`_ or `issue tracker`_.
+
+.. _discussion board: https://github.com/PGScatalog/pgsc_calc/discussions
+.. _issue tracker: https://github.com/pgscatalog/pgsc_calc/issues
Citations
~~~~~~~~~
If you use ``pgscatalog/pgsc_calc`` in your analysis, please cite:
- PGS Catalog Calculator `(in development)`. PGS Catalog
+ PGS Catalog Calculator [0]_. PGS Catalog
Team. https://github.com/PGScatalog/pgsc_calc
Lambert `et al.` (2021) The Polygenic Score Catalog as an open database for
@@ -167,6 +175,8 @@ you use in your analyses, and the underlying data/software tools described in th
.. _citations file: https://github.com/PGScatalog/pgsc_calc/blob/master/CITATIONS.md
.. _10.1038/s41588-021-00783-5: https://doi.org/10.1038/s41588-021-00783-5
+.. [0] A manuscript is in development but the calculated scores have been
+ for consistency in UK Biobank since v1.1.0
License Information
diff --git a/docs/reference/params.rst b/docs/reference/params.rst
index 9110a87c..c982fe6c 100644
--- a/docs/reference/params.rst
+++ b/docs/reference/params.rst
@@ -1,7 +1,7 @@
.. _param ref:
-Reference: Input Parameters/Flags
-=================================
+Reference: Workflow parameters
+==============================
The documentation below is automatically generated from the input schema and
contains additional technical detail. **Parameters in bold** are required and
diff --git a/nextflow_schema.json b/nextflow_schema.json
index af4ac1df..975dd273 100644
--- a/nextflow_schema.json
+++ b/nextflow_schema.json
@@ -1,422 +1,455 @@
{
- "$schema": "http://json-schema.org/draft-07/schema",
- "$id": "https://raw.githubusercontent.com/pgscatalog/pgsc_calc/master/nextflow_schema.json",
- "title": "pgscatalog/pgsc_calc pipeline parameters",
- "description": "This pipeline applies scoring files from the PGS Catalog to target set(s) of genotyped samples",
- "type": "object",
- "definitions": {
- "input_output_options": {
- "title": "Input/output options",
- "type": "object",
- "fa_icon": "fas fa-terminal",
- "description": "Define where the pipeline should find input data and save output data.",
- "properties": {
- "input": {
- "type": "string",
- "description": "Path to input samplesheet",
- "format": "file-path"
- },
- "format": {
- "type": "string",
- "default": "csv",
- "fa_icon": "fas fa-cog",
- "description": "Format of input samplesheet",
- "enum": ["csv", "json"]
- },
- "scorefile": {
- "type": "string",
- "description": "Path to a scoring file in PGS Catalog format. Multiple scorefiles can be specified using wildcards (e.g., ``--scorefile \"path/to/scores/*.txt\"``)",
- "fa_icon": "fas fa-file-alt",
- "format": "file-path"
- },
- "pgs_id": {
- "type": "string",
- "description": "A comma separated list of PGS score IDs, e.g. PGS000802"
- },
- "pgp_id": {
- "type": "string",
- "description": "A comma separated list of PGS Catalog publications, e.g. PGP000001"
- },
- "trait_efo": {
- "type": "string",
- "description": "A comma separated list of PGS Catalog EFO traits, e.g. EFO_0004214"
- },
- "efo_direct": {
- "type": "boolean",
- "description": "Return only PGS tagged with exact EFO term (e.g. no PGS for child/descendant terms in the ontology)"
- },
- "copy_genomes": {
- "type": "boolean",
- "description": "Copy harmonised genomes (plink2 pgen/pvar/psam files) to outdir"
- },
- "genotypes_cache": {
- "type": "string",
- "default": "None",
- "description": "A path to a directory that should contain relabelled genotypes",
- "format": "directory-path"
- },
- "outdir": {
- "type": "string",
- "description": "Path to the output directory where the results will be saved.",
- "default": "./results",
- "fa_icon": "fas fa-folder-open",
- "format": "directory-path"
- },
- "email": {
- "type": "string",
- "description": "Email address for completion summary.",
- "fa_icon": "fas fa-envelope",
- "help_text": "Set this parameter to your e-mail address to get a summary e-mail with details of the run sent to you when the workflow exits. If set in your user config file (`~/.nextflow/config`) then you don't need to specify this on the command line for every run.",
- "pattern": "^([a-zA-Z0-9_\\-\\.]+)@([a-zA-Z0-9_\\-\\.]+)\\.([a-zA-Z]{2,5})$"
- }
- },
- "required": ["input", "format"]
- },
- "ancestry_options": {
- "title": "Ancestry options",
- "type": "object",
- "description": "",
- "default": "",
- "properties": {
- "ancestry_params_file": {
- "type": "string",
- "default": "None",
- "description": "A YAML or JSON file that contains parameters for handling ancestry options"
- },
- "projection_method": {
- "type": "string",
- "default": "oadp",
- "enum": ["oadp", "sp", "adp"],
- "description": "The method for PCA prediction. oadp: most accurate. adp: accurate but slow. sp: fast but inaccurate."
- },
- "ancestry_method": {
- "type": "string",
- "default": "RandomForest",
- "description": "Method used for population/ancestry assignment",
- "enum": ["Mahalanobis", "RandomForest"]
- },
- "ref_label": {
- "type": "string",
- "default": "SuperPop",
- "description": "Population labels in reference psam to use for assignment"
- },
- "n_popcomp": {
- "type": "integer",
- "default": 10,
- "description": "Number of PCs used for population assignment"
- },
- "normalization_method": {
- "type": "string",
- "default": "empirical mean mean+var",
- "description": "Method used for normalisation of genetic ancestry",
- "enum": ["empirical", "mean", "mean+var", "empirical mean mean+var"]
- },
- "n_normalization": {
- "type": "integer",
- "default": 4,
- "description": "Number of PCs used for population normalisation"
- },
- "load_afreq": {
- "type": "boolean",
- "default": true,
- "description": "Load allelic frequencies from reference panel when scoring target genomes"
- }
- },
- "required": [
- "projection_method",
- "ancestry_method",
- "ref_label",
- "n_popcomp",
- "normalization_method",
- "n_normalization",
- "load_afreq"
- ]
- },
- "reference_options": {
- "title": "Reference options",
- "type": "object",
- "description": "Define how reference genomes are defined and processed",
- "default": "",
- "properties": {
- "run_ancestry": {
- "type": "string",
- "default": "None",
- "format": "file-path",
- "description": "Path to reference database. Must be set if --ref_samplesheet is not set."
- },
- "ref_samplesheet": {
- "type": "string",
- "description": "Path to a samplesheet that describes the structure of reference data. Must be set if --ref isn't set."
- },
- "hg19_chain": {
- "type": "string",
- "default": "https://hgdownload.cse.ucsc.edu/goldenpath/hg19/liftOver/hg19ToHg38.over.chain.gz",
- "description": "Path to a UCSC chain file for converting from hg19 to hg38",
- "pattern": ".*chain.gz$",
- "format": "file-path"
- },
- "hg38_chain": {
- "type": "string",
- "default": "https://hgdownload.soe.ucsc.edu/goldenPath/hg38/liftOver/hg38ToHg19.over.chain.gz",
- "description": "Path to a UCSC chain file for converting from hg38 to hg19"
- },
- "geno_ref": {
- "type": "number",
- "default": 0.1,
- "description": "Exclude variants with missing call frequencies greater than a threshold (in reference genomes)",
- "minimum": 0,
- "maximum": 1
- },
- "mind_ref": {
- "type": "number",
- "default": 0.1,
- "description": "Exclude samples with missing call frequencies greater than a threshold (in reference genomes)",
- "minimum": 0,
- "maximum": 1
- },
- "maf_ref": {
- "type": "number",
- "default": 0.01,
- "description": "Exclude variants with allele frequency lower than a threshold (in reference genomes)",
- "minimum": 0,
- "maximum": 1
- },
- "hwe_ref": {
- "type": "number",
- "default": 1e-6,
- "description": "Exclude variants with Hardy-Weinberg equilibrium exact test p-values below a threshold (in reference genomes)",
- "minimum": 0,
- "maximum": 1
- },
- "indep_pairwise_ref": {
- "type": "string",
- "default": "1000 50 0.05",
- "description": "Used to generate a list of variants in approximate linkage equilibrium in reference genomes. Window size - step size - unphased hardcall r^2 threshold."
- },
- "ld_grch37": {
- "type": "string",
- "description": "Path to a file that contains areas of high linkage disequilibrium in the reference data (build GRCh37).",
- "format": "file-path",
- "mimetype": "text/plain"
- },
- "ld_grch38": {
- "type": "string",
- "description": "Path to a file that contains areas of high linkage disequilibrium in the reference data (build GRCh38).",
- "format": "file-path",
- "mimetype": "text/plain"
+ "$schema": "http://json-schema.org/draft-07/schema",
+ "$id": "https://raw.githubusercontent.com/pgscatalog/pgsc_calc/master/nextflow_schema.json",
+ "title": "pgscatalog/pgsc_calc pipeline parameters",
+ "description": "This pipeline applies scoring files from the PGS Catalog to target set(s) of genotyped samples",
+ "type": "object",
+ "definitions": {
+ "input_output_options": {
+ "title": "Input/output options",
+ "type": "object",
+ "fa_icon": "fas fa-terminal",
+ "description": "Define where the pipeline should find input data and save output data.",
+ "properties": {
+ "input": {
+ "type": "string",
+ "description": "Path to input samplesheet",
+ "format": "file-path"
+ },
+ "format": {
+ "type": "string",
+ "default": "csv",
+ "fa_icon": "fas fa-cog",
+ "description": "Format of input samplesheet",
+ "enum": [
+ "csv",
+ "json"
+ ]
+ },
+ "scorefile": {
+ "type": "string",
+ "description": "Path to a scoring file in PGS Catalog format. Multiple scorefiles can be specified using wildcards (e.g., ``--scorefile \"path/to/scores/*.txt\"``)",
+ "fa_icon": "fas fa-file-alt",
+ "format": "file-path"
+ },
+ "pgs_id": {
+ "type": "string",
+ "description": "A comma separated list of PGS score IDs, e.g. PGS000802"
+ },
+ "pgp_id": {
+ "type": "string",
+ "description": "A comma separated list of PGS Catalog publications, e.g. PGP000001"
+ },
+ "trait_efo": {
+ "type": "string",
+ "description": "A comma separated list of PGS Catalog EFO traits, e.g. EFO_0004214"
+ },
+ "efo_direct": {
+ "type": "boolean",
+ "description": "Return only PGS tagged with exact EFO term (e.g. no PGS for child/descendant terms in the ontology)"
+ },
+ "copy_genomes": {
+ "type": "boolean",
+ "description": "Copy harmonised genomes (plink2 pgen/pvar/psam files) to outdir"
+ },
+ "genotypes_cache": {
+ "type": "string",
+ "default": "None",
+ "description": "A path to a directory that should contain relabelled genotypes",
+ "format": "directory-path"
+ },
+ "outdir": {
+ "type": "string",
+ "description": "Path to the output directory where the results will be saved.",
+ "default": "./results",
+ "fa_icon": "fas fa-folder-open",
+ "format": "directory-path"
+ },
+ "email": {
+ "type": "string",
+ "description": "Email address for completion summary.",
+ "fa_icon": "fas fa-envelope",
+ "help_text": "Set this parameter to your e-mail address to get a summary e-mail with details of the run sent to you when the workflow exits. If set in your user config file (`~/.nextflow/config`) then you don't need to specify this on the command line for every run.",
+ "pattern": "^([a-zA-Z0-9_\\-\\.]+)@([a-zA-Z0-9_\\-\\.]+)\\.([a-zA-Z]{2,5})$"
+ }
+ },
+ "required": [
+ "input",
+ "format"
+ ]
+ },
+ "ancestry_options": {
+ "title": "Ancestry options",
+ "type": "object",
+ "description": "",
+ "default": "",
+ "properties": {
+ "ancestry_params_file": {
+ "type": "string",
+ "default": "None",
+ "description": "A YAML or JSON file that contains parameters for handling ancestry options"
+ },
+ "projection_method": {
+ "type": "string",
+ "default": "oadp",
+ "enum": [
+ "oadp",
+ "sp",
+ "adp"
+ ],
+ "description": "The method for PCA prediction. oadp: most accurate. adp: accurate but slow. sp: fast but inaccurate."
+ },
+ "ancestry_method": {
+ "type": "string",
+ "default": "RandomForest",
+ "description": "Method used for population/ancestry assignment",
+ "enum": [
+ "Mahalanobis",
+ "RandomForest"
+ ]
+ },
+ "ref_label": {
+ "type": "string",
+ "default": "SuperPop",
+ "description": "Population labels in reference psam to use for assignment"
+ },
+ "n_popcomp": {
+ "type": "integer",
+ "default": 5,
+ "description": "Number of PCs used for population assignment"
+ },
+ "normalization_method": {
+ "type": "string",
+ "default": "empirical mean mean+var",
+ "description": "Method used for normalisation of genetic ancestry",
+ "enum": [
+ "empirical",
+ "mean",
+ "mean+var",
+ "empirical mean mean+var"
+ ]
+ },
+ "n_normalization": {
+ "type": "integer",
+ "default": 4,
+ "description": "Number of PCs used for population normalisation"
+ },
+ "load_afreq": {
+ "type": "boolean",
+ "default": true,
+ "description": "Load allelic frequencies from reference panel when scoring target genomes"
+ }
+ },
+ "required": [
+ "projection_method",
+ "ancestry_method",
+ "ref_label",
+ "n_popcomp",
+ "normalization_method",
+ "n_normalization",
+ "load_afreq"
+ ]
+ },
+ "reference_options": {
+ "title": "Reference options",
+ "type": "object",
+ "description": "Define how reference genomes are defined and processed",
+ "default": "",
+ "properties": {
+ "run_ancestry": {
+ "type": "string",
+ "default": "None",
+ "format": "file-path",
+ "description": "Path to reference database. Must be set if --ref_samplesheet is not set."
+ },
+ "ref_samplesheet": {
+ "type": "string",
+ "description": "Path to a samplesheet that describes the structure of reference data. Must be set if --ref isn't set."
+ },
+ "hg19_chain": {
+ "type": "string",
+ "description": "Path to a UCSC chain file for converting from hg19 to hg38. Needed if lifting over a custom scoring file.",
+ "pattern": ".*chain.gz$",
+ "format": "file-path",
+ "mimetype": "application/gzip"
+ },
+ "hg38_chain": {
+ "type": "string",
+ "description": "Path to a UCSC chain file for converting from hg38 to hg19. Needed if lifting over a custom scoring file.",
+ "format": "file-path",
+ "mimetype": "application/gzip"
+ },
+ "geno_ref": {
+ "type": "number",
+ "default": 0.1,
+ "description": "Exclude variants with missing call frequencies greater than a threshold (in reference genomes)",
+ "minimum": 0,
+ "maximum": 1
+ },
+ "mind_ref": {
+ "type": "number",
+ "default": 0.1,
+ "description": "Exclude samples with missing call frequencies greater than a threshold (in reference genomes)",
+ "minimum": 0,
+ "maximum": 1
+ },
+ "maf_ref": {
+ "type": "number",
+ "default": 0.05,
+ "description": "Exclude variants with allele frequency lower than a threshold (in reference genomes)",
+ "minimum": 0,
+ "maximum": 1
+ },
+ "hwe_ref": {
+ "type": "number",
+ "default": 1e-04,
+ "description": "Exclude variants with Hardy-Weinberg equilibrium exact test p-values below a threshold (in reference genomes)",
+ "minimum": 0,
+ "maximum": 1
+ },
+ "indep_pairwise_ref": {
+ "type": "string",
+ "default": "1000 50 0.05",
+ "description": "Used to generate a list of variants in approximate linkage equilibrium in reference genomes. Window size - step size - unphased hardcall r^2 threshold."
+ },
+ "ld_grch37": {
+ "type": "string",
+ "description": "Path to a file that contains areas of high linkage disequilibrium in the reference data (build GRCh37).",
+ "format": "file-path",
+ "mimetype": "text/plain"
+ },
+ "ld_grch38": {
+ "type": "string",
+ "description": "Path to a file that contains areas of high linkage disequilibrium in the reference data (build GRCh38).",
+ "format": "file-path",
+ "mimetype": "text/plain"
+ }
+ },
+ "required": [
+ "geno_ref",
+ "mind_ref",
+ "maf_ref",
+ "hwe_ref",
+ "indep_pairwise_ref",
+ "ld_grch37",
+ "ld_grch38"
+ ]
+ },
+ "compatibility_options": {
+ "title": "Compatibility options",
+ "type": "object",
+ "description": "Define parameters that control how scoring files and target genomes are made compatible with each other",
+ "default": "",
+ "properties": {
+ "compat_params_file": {
+ "type": "string",
+ "default": "None",
+ "description": "A YAML or JSON file that contains parameters for handling compatibility options"
+ },
+ "target_build": {
+ "type": "string",
+ "enum": [
+ "GRCh37",
+ "GRCh38"
+ ],
+ "description": "Genome build of target genomes"
+ },
+ "liftover": {
+ "type": "boolean",
+ "description": "Lift scoring files to match your target genomes. Requires build information in the header of the scoring files."
+ },
+ "min_lift": {
+ "type": "number",
+ "default": 0.95,
+ "description": "Minimum proportion of variants required to successfully remap a scoring file to a different genome build",
+ "minimum": 0,
+ "maximum": 1
+ }
+ },
+ "required": [
+ "target_build"
+ ]
+ },
+ "matching_options": {
+ "title": "Matching options",
+ "type": "object",
+ "description": "Define how variants are matched across scoring files and target genomes",
+ "default": "",
+ "properties": {
+ "keep_multiallelic": {
+ "type": "boolean",
+ "description": "Allow matches of scoring file variants to multiallelic variants in the target dataset"
+ },
+ "keep_ambiguous": {
+ "type": "boolean",
+ "description": "Keep matches of scoring file variants to strand ambiguous variants (e.g. A/T and C/G SNPs) in the target dataset. This assumes the scoring file and target dataset report variants on the same strand."
+ },
+ "fast_match": {
+ "type": "boolean",
+ "description": "Enable fast matching, which significantly increases RAM usage (32GB minimum recommended)"
+ },
+ "min_overlap": {
+ "type": "number",
+ "default": 0.75,
+ "description": "Minimum proportion of variants present in both the score file and input target genomic data",
+ "fa_icon": "fas fa-cog",
+ "minimum": 0,
+ "maximum": 1
+ }
+ },
+ "fa_icon": "fas fa-user-cog"
+ },
+ "max_job_request_options": {
+ "title": "Max job request options",
+ "type": "object",
+ "fa_icon": "fab fa-acquisitions-incorporated",
+ "description": "Set the top limit for requested resources for any single job.",
+ "help_text": "If you are running on a smaller system, a pipeline step requesting more resources than are available may cause the Nextflow to stop the run with an error. These options allow you to cap the maximum resources requested by any single job so that the pipeline will run on your system.\n\nNote that you can not _increase_ the resources requested by any job using these options. For that you will need your own configuration file. See [the nf-core website](https://nf-co.re/usage/configuration) for details.",
+ "properties": {
+ "max_cpus": {
+ "type": "integer",
+ "description": "Maximum number of CPUs that can be requested for any single job.",
+ "default": 16,
+ "fa_icon": "fas fa-microchip",
+ "hidden": true,
+ "help_text": "Use to set an upper-limit for the CPU requirement for each process. Should be an integer e.g. `--max_cpus 1`"
+ },
+ "max_memory": {
+ "type": "string",
+ "description": "Maximum amount of memory that can be requested for any single job.",
+ "default": "128.GB",
+ "fa_icon": "fas fa-memory",
+ "pattern": "^\\d+(\\.\\d+)?\\.?\\s*(K|M|G|T)?B$",
+ "hidden": true,
+ "help_text": "Use to set an upper-limit for the memory requirement for each process. Should be a string in the format integer-unit e.g. `--max_memory '8.GB'`"
+ },
+ "max_time": {
+ "type": "string",
+ "description": "Maximum amount of time that can be requested for any single job.",
+ "default": "240.h",
+ "fa_icon": "far fa-clock",
+ "pattern": "^(\\d+\\.?\\s*(s|m|h|day)\\s*)+$",
+ "hidden": true,
+ "help_text": "Use to set an upper-limit for the time requirement for each process. Should be a string in the format integer-unit e.g. `--max_time '2.h'`"
+ }
+ }
+ },
+ "generic_options": {
+ "title": "Generic options",
+ "type": "object",
+ "fa_icon": "fas fa-file-import",
+ "description": "Less common options for the pipeline, typically set in a config file.",
+ "help_text": "These options are common to all nf-core pipelines and allow you to customise some of the core preferences for how the pipeline runs.\n\nTypically these options would be set in a Nextflow config file loaded for all pipeline runs, such as `~/.nextflow/config`.",
+ "properties": {
+ "help": {
+ "type": "boolean",
+ "description": "Display help text.",
+ "fa_icon": "fas fa-question-circle",
+ "hidden": true
+ },
+ "publish_dir_mode": {
+ "type": "string",
+ "default": "copy",
+ "description": "Method used to save pipeline results to output directory.",
+ "help_text": "The Nextflow `publishDir` option specifies which intermediate files should be saved to the output directory. This option tells the pipeline what method should be used to move these files. See [Nextflow docs](https://www.nextflow.io/docs/latest/process.html#publishdir) for details.",
+ "fa_icon": "fas fa-copy",
+ "enum": [
+ "symlink",
+ "rellink",
+ "link",
+ "copy",
+ "copyNoFollow",
+ "move"
+ ],
+ "hidden": true
+ },
+ "email_on_fail": {
+ "type": "string",
+ "description": "Email address for completion summary, only when pipeline fails.",
+ "fa_icon": "fas fa-exclamation-triangle",
+ "pattern": "^([a-zA-Z0-9_\\-\\.]+)@([a-zA-Z0-9_\\-\\.]+)\\.([a-zA-Z]{2,5})$",
+ "help_text": "An email address to send a summary email to when the pipeline is completed - ONLY sent if the pipeline does not exit successfully.",
+ "hidden": true
+ },
+ "plaintext_email": {
+ "type": "boolean",
+ "description": "Send plain-text email instead of HTML.",
+ "fa_icon": "fas fa-remove-format",
+ "hidden": true
+ },
+ "monochrome_logs": {
+ "type": "boolean",
+ "description": "Do not use coloured log outputs.",
+ "fa_icon": "fas fa-palette",
+ "hidden": true
+ },
+ "tracedir": {
+ "type": "string",
+ "description": "Directory to keep pipeline Nextflow logs and reports.",
+ "default": "${params.outdir}/pipeline_info",
+ "fa_icon": "fas fa-cogs",
+ "hidden": true
+ },
+ "validate_params": {
+ "type": "boolean",
+ "description": "Boolean whether to validate parameters against the schema at runtime",
+ "default": true,
+ "fa_icon": "fas fa-check-square",
+ "hidden": true
+ },
+ "show_hidden_params": {
+ "type": "boolean",
+ "fa_icon": "far fa-eye-slash",
+ "description": "Show all params when using `--help`",
+ "hidden": true,
+ "help_text": "By default, parameters set as _hidden_ in the schema are not shown on the command line when a user runs with `--help`. Specifying this option will tell the pipeline to show all parameters."
+ },
+ "platform": {
+ "type": "string",
+ "default": "amd64",
+ "enum": [
+ "amd64",
+ "arm64"
+ ],
+ "description": "What platform is the pipeline executing on?"
+ },
+ "parallel": {
+ "type": "boolean",
+ "description": "Enable parallel calculation of scores. This is I/O and RAM intensive."
+ }
+ },
+ "required": [
+ "platform"
+ ]
}
- },
- "required": [
- "hg19_chain",
- "hg38_chain",
- "geno_ref",
- "mind_ref",
- "maf_ref",
- "hwe_ref",
- "indep_pairwise_ref",
- "ld_grch37",
- "ld_grch38"
- ]
},
- "compatibility_options": {
- "title": "Compatibility options",
- "type": "object",
- "description": "Define parameters that control how scoring files and target genomes are made compatible with each other",
- "default": "",
- "properties": {
- "compat_params_file": {
- "type": "string",
- "default": "None",
- "description": "A YAML or JSON file that contains parameters for handling compatibility options"
+ "allOf": [
+ {
+ "$ref": "#/definitions/input_output_options"
},
- "target_build": {
- "type": "string",
- "enum": ["GRCh37", "GRCh38"],
- "description": "Genome build of target genomes"
+ {
+ "$ref": "#/definitions/ancestry_options"
},
- "liftover": {
- "type": "boolean",
- "description": "Lift scoring files to match your target genomes. Requires build information in the header of the scoring files."
+ {
+ "$ref": "#/definitions/reference_options"
},
- "min_lift": {
- "type": "number",
- "default": 0.95,
- "description": "Minimum proportion of variants required to successfully remap a scoring file to a different genome build",
- "minimum": 0,
- "maximum": 1
- }
- },
- "required": ["target_build"]
- },
- "matching_options": {
- "title": "Matching options",
- "type": "object",
- "description": "Define how variants are matched across scoring files and target genomes",
- "default": "",
- "properties": {
- "keep_multiallelic": {
- "type": "boolean",
- "description": "Allow matches of scoring file variants to multiallelic variants in the target dataset"
+ {
+ "$ref": "#/definitions/compatibility_options"
},
- "keep_ambiguous": {
- "type": "boolean",
- "description": "Keep matches of scoring file variants to strand ambiguous variants (e.g. A/T and C/G SNPs) in the target dataset. This assumes the scoring file and target dataset report variants on the same strand."
+ {
+ "$ref": "#/definitions/matching_options"
},
- "fast_match": {
- "type": "boolean",
- "description": "Enable fast matching, which significantly increases RAM usage (32GB minimum recommended)"
+ {
+ "$ref": "#/definitions/max_job_request_options"
},
- "min_overlap": {
- "type": "number",
- "default": 0.75,
- "description": "Minimum proportion of variants present in both the score file and input target genomic data",
- "fa_icon": "fas fa-cog",
- "minimum": 0,
- "maximum": 1
+ {
+ "$ref": "#/definitions/generic_options"
}
- },
- "fa_icon": "fas fa-user-cog"
- },
- "max_job_request_options": {
- "title": "Max job request options",
- "type": "object",
- "fa_icon": "fab fa-acquisitions-incorporated",
- "description": "Set the top limit for requested resources for any single job.",
- "help_text": "If you are running on a smaller system, a pipeline step requesting more resources than are available may cause the Nextflow to stop the run with an error. These options allow you to cap the maximum resources requested by any single job so that the pipeline will run on your system.\n\nNote that you can not _increase_ the resources requested by any job using these options. For that you will need your own configuration file. See [the nf-core website](https://nf-co.re/usage/configuration) for details.",
- "properties": {
- "max_cpus": {
- "type": "integer",
- "description": "Maximum number of CPUs that can be requested for any single job.",
- "default": 16,
- "fa_icon": "fas fa-microchip",
- "hidden": true,
- "help_text": "Use to set an upper-limit for the CPU requirement for each process. Should be an integer e.g. `--max_cpus 1`"
- },
- "max_memory": {
- "type": "string",
- "description": "Maximum amount of memory that can be requested for any single job.",
- "default": "128.GB",
- "fa_icon": "fas fa-memory",
- "pattern": "^\\d+(\\.\\d+)?\\.?\\s*(K|M|G|T)?B$",
- "hidden": true,
- "help_text": "Use to set an upper-limit for the memory requirement for each process. Should be a string in the format integer-unit e.g. `--max_memory '8.GB'`"
- },
- "max_time": {
- "type": "string",
- "description": "Maximum amount of time that can be requested for any single job.",
- "default": "240.h",
- "fa_icon": "far fa-clock",
- "pattern": "^(\\d+\\.?\\s*(s|m|h|day)\\s*)+$",
- "hidden": true,
- "help_text": "Use to set an upper-limit for the time requirement for each process. Should be a string in the format integer-unit e.g. `--max_time '2.h'`"
+ ],
+ "properties": {
+ "ancestry_checksums": {
+ "type": "string",
+ "default": "/Users/bwingfield/Documents/projects/pgsc_calc/assets/ancestry/checksums.txt"
}
- }
- },
- "generic_options": {
- "title": "Generic options",
- "type": "object",
- "fa_icon": "fas fa-file-import",
- "description": "Less common options for the pipeline, typically set in a config file.",
- "help_text": "These options are common to all nf-core pipelines and allow you to customise some of the core preferences for how the pipeline runs.\n\nTypically these options would be set in a Nextflow config file loaded for all pipeline runs, such as `~/.nextflow/config`.",
- "properties": {
- "help": {
- "type": "boolean",
- "description": "Display help text.",
- "fa_icon": "fas fa-question-circle",
- "hidden": true
- },
- "publish_dir_mode": {
- "type": "string",
- "default": "copy",
- "description": "Method used to save pipeline results to output directory.",
- "help_text": "The Nextflow `publishDir` option specifies which intermediate files should be saved to the output directory. This option tells the pipeline what method should be used to move these files. See [Nextflow docs](https://www.nextflow.io/docs/latest/process.html#publishdir) for details.",
- "fa_icon": "fas fa-copy",
- "enum": [
- "symlink",
- "rellink",
- "link",
- "copy",
- "copyNoFollow",
- "move"
- ],
- "hidden": true
- },
- "email_on_fail": {
- "type": "string",
- "description": "Email address for completion summary, only when pipeline fails.",
- "fa_icon": "fas fa-exclamation-triangle",
- "pattern": "^([a-zA-Z0-9_\\-\\.]+)@([a-zA-Z0-9_\\-\\.]+)\\.([a-zA-Z]{2,5})$",
- "help_text": "An email address to send a summary email to when the pipeline is completed - ONLY sent if the pipeline does not exit successfully.",
- "hidden": true
- },
- "plaintext_email": {
- "type": "boolean",
- "description": "Send plain-text email instead of HTML.",
- "fa_icon": "fas fa-remove-format",
- "hidden": true
- },
- "monochrome_logs": {
- "type": "boolean",
- "description": "Do not use coloured log outputs.",
- "fa_icon": "fas fa-palette",
- "hidden": true
- },
- "tracedir": {
- "type": "string",
- "description": "Directory to keep pipeline Nextflow logs and reports.",
- "default": "${params.outdir}/pipeline_info",
- "fa_icon": "fas fa-cogs",
- "hidden": true
- },
- "validate_params": {
- "type": "boolean",
- "description": "Boolean whether to validate parameters against the schema at runtime",
- "default": true,
- "fa_icon": "fas fa-check-square",
- "hidden": true
- },
- "show_hidden_params": {
- "type": "boolean",
- "fa_icon": "far fa-eye-slash",
- "description": "Show all params when using `--help`",
- "hidden": true,
- "help_text": "By default, parameters set as _hidden_ in the schema are not shown on the command line when a user runs with `--help`. Specifying this option will tell the pipeline to show all parameters."
- },
- "platform": {
- "type": "string",
- "default": "amd64",
- "enum": ["amd64", "arm64"],
- "description": "What platform is the pipeline executing on?"
- },
- "parallel": {
- "type": "boolean",
- "description": "Enable parallel calculation of scores. This is I/O and RAM intensive."
- }
- },
- "required": ["platform"]
- }
- },
- "allOf": [
- {
- "$ref": "#/definitions/input_output_options"
- },
- {
- "$ref": "#/definitions/ancestry_options"
- },
- {
- "$ref": "#/definitions/reference_options"
- },
- {
- "$ref": "#/definitions/compatibility_options"
- },
- {
- "$ref": "#/definitions/matching_options"
- },
- {
- "$ref": "#/definitions/max_job_request_options"
- },
- {
- "$ref": "#/definitions/generic_options"
}
- ]
}