Sacurine statistical workflow
By Etienne Thévenot (last updated: 2017-08-15)
- Univariate and multivariate analysis of metabolomics data
- The "W4M univariate-multivariate-statistics" history (W4M00001)
- Tutorial
- Interpretation of the results
- Enjoy your analyses!
- Acknowledgements
Statistics are a critical step in metabolomics (and the other omics) data analysis, e.g. to:
- explore the dataset (visualize the data, detect outlier samples or variables, identify cluster of samples)
- identify which variables have intensities signficantly distinct between levels of qualitative factor (or are signficantly correlated with a quantitative response)
- build predictive models of these responses
- select variables which significantly contribute to the performance of these models
Three modules have been developed in Galaxy to perform such statistics:
- univariate for step 2
- multivariate (Wold, 2001; Trygg and Wold, 2002; Thévenot et al, 2015) for steps 1 (Principal Component Analysis) and 3 (Partial Least-Squares)
- biosigner (Rinaudo et al, 2016) for step 4 (significant signatures for either Partial Least Square Discriminant Analysis, Random Forest, or Support Vector Machine classifiers between two groups). They are available on the Workflow4Metabolomics e-infrastructure (Guitton et al, 2017), and also on the PhenoMeNal instance as described in the tutorial below.
These analyses take a input a sample by variable table of intensities (e.g. an MS peak table, or an NMR table of bucket intensities) in addition to, for steps 2-3, the values of the response (e.g. treatment, body mass index, etc.). They return metrics about samples (e.g. scores) or variables (e.g. p-values, loadings, Variable Importance in Projection).
This history, which is publicly available on the PhenoMeNal instance, is an example of a statistical workflow applied to metabolomics data. The objective is to characterize the physiological variations of the urine metabolome with age, body mass index (BMI), and gender in a human cohort (Thévenot et al, 2015).
The dataset (named sacurine and available from the MetaboLights repository: MTBLS404) consists of:
- a peak table of 183 samples and 110 metabolites
- the age, BMI, and gender of the 183 volunteers
The format of the dataset consists of 3 tables: the dataMatrix, the sampleMetadata, and the variableMetadata tabulated files (.tsv files which you can open with spreadsheet software such as Excel or OpenOffice). More details about these formats can be found on slides 4-9 from this presentation.
The workflow (W4M00001) consists of:
- Principal Component Analysis (PCA) of the dataset
- Student's t-test of intensity differences between gender, with correction for multiple testing (False Discovery Rate, FDR)
- Orthogonal Partial Least Squares - Discriminant Analysis (OPLS-DA) modeling of gender
- Selection of significant signatures for prediction with either Partial Least Squares - Discriminant Analysis (PLS-DA), Random Forest, or Support Vector Machine (SVM).
Note: More details about this history can be found on the Workflow4Metabolomics platform (W4M00001; DOI:10.15454/1.4811121736910142E12).
The purpose of this tutorial is to show you how to import and re-run the history, to better understand how to use the statistical modules for your own analyses.
The history (i.e., the workflow and associated input and ouput data) is available at our PhenoMeNal public instance.
The tutorial (which you can follow either with the videos or with the accompanying text) shows you how to:
- import the history into your own account,
- extract the workflow,
- modify the workflow input parameters (optionally),
- and run it with either the example data or your own.
Note: if you are new to Galaxy we strongly advise you to first read the "Galaxy 101 - What is Galaxy?" tutorial which will help you get a better understanding about the basic concepts when running workflows in Galaxy.
The complete workflow can be viewed in action by watching the following two video tutorials:
This video tutorial explains the procedure to load the workflow and match the loaded data to the inputs of the workflow.
On the welcome page of Galaxy you will find a menu item "Login or register". Please make sure you are logged in before proceeding to step two. If you don't have an account yet, you can use the registration form to request access. If you proceed without an account you will not be able to extract the workflows and re-run it, you will only be able to view the results of the shared history.

In the same menu at the top of the page you will find an option "Shared Data". Here you are able to load the history of this tutorial by going to the "Histories" option of the drop-down menu. This will bring you to an overview of histories shared by others.

For this tutorial you have to select the "Univariate and Multivariate Statistics" history. After you click on the link it will give an overview of the dataset.

If all went well, you should see a list of intermediate datasets, all shown on a green background. The last thing we have to do here is click on the "import history" link it the upper right part of the page. This will give you the option to rename the history into something else, or leave it as "imported: ...". Finally click on "Import" to save the history in your own account.

When done correctly, you should see the history appear in the right part of the screen. If so, you can proceed to the next step.
By now you can view the history, but to run the actual workflow you have to extract it first from this imported history. This is done by clicking on the "History options" button (wheel/gear icon) in the upper right part of the screen, right above the search box. Select the "Extract Workflow" option here.

Same as for the history, you can now change the name of the workflow, and click on the "Create Workflow" button below the name to extract and save the workflow to your account. When done correctly, you should now see a blue bar with some text that allows you to edit or run the workflow.

If so, you can proceed to the next step.
You are now able to view, edit and run the extracted workflow. In the menu at the top of the page you find an option "Workflow". On that page you can select the extracted workflow from step 3. When you choose the edit option you can change the individual tools and the parameters used when running the workflow. For example you could change the "(Corrected) p-value significance threshold" from 0.05 to 0.01 in the Univariate tool to get more stringent results.

After you make changes to the workflow you have to save them before leaving the "edit" page. This is done by clicking the "Settings" button (wheel/gear icon) in the upper right part of the screen.
Warning: Saving workflows is very important since, unlike histories, workflows are not saved automatically.
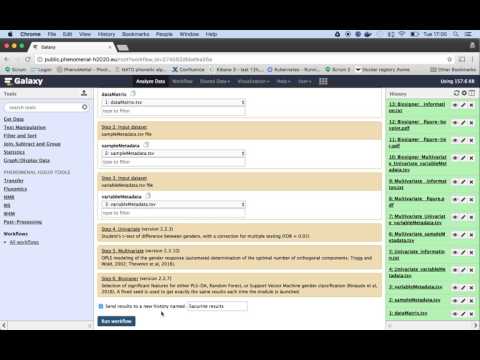
Same as step 4, in the menu at the top of the page you find an option "Workflow". On that page you can select the edited workflow from step 4. But now instead of selecting "edit", we choose "run". This page will allow you to overwrite or create a new history, and select the required input data. Choose a new history, and make sure the "dataMatrix.tsv" is selected in the first input field, "sampleMetaData" in the second, and "variableMetaData.tsv" in the third. When this is done you can click on "Run workflow", a blue button on the top of the page.

The tools of the workflow will now be scheduled to run as soon as resource become available. In case you selected "Send results to a new history", you will have to switch histories first! This is done using the "View all histories" button in the upper right corner, next to the "History options" button from step 3. Here you can select the correct (newly created) history. You can now follow the progress in the "history" pane on the right. When all boxed are green it finished. You can now view the result of the individual tools by clicking on the "eye"-icon of the appropriate tool.

Now that we have successfully ran an imported history and workflow, we can look at the results. All tools with output will show an "eye"-icon in the green box of the workflow history. This will open/preview the results in Galaxy. Optionally you can edit, download or delete results.

Below is a description of some of the ouputs from each module. You can find additional details about the statistical modules (parameters, diagnostics, output files and graphics) in:
- the documentation section at the bottom of the Galaxy pages (where you select your parameters)
- the course presentations on the Workflow4Metabolomics plaform about univariate and multivariate statistics
- the vignettes from the ropls and biosigner packages
The defaults graphic is shown below (as we are more interested in the gender factor here, we asked to color the points from the score plots by setting Ellipses to gender, in the Advanced graphical parameters). We observe that the difference between genders is not main source of variance in the first two PCA dimensions.
 PCA
PCA summary plot. Top left overview: the scree plot (i.e., inertia barplot) suggests that 3 components may be sufficient to capture most of the inertia; Top right outlier: this graphics shows the distances within
and orthogonal to the projection plane (see Hubert et al., 2005): the name of the samples with a high value for at least one of the distances are indicated; Bottom left x-score: the variance along each axis equals the variance captured by each component: it therefore depends on the total variance of the dataMatrix X and of the percentage of this variance captured by the component (indicated in the labels); it decreases when going from one component to a component with higher indice; Bottom right x-loading: the 3 variables with most extreme
values (positive and negative) for each loading are black colored and labeled.
Note:
-
A specific
Graphic typecan be selected within theAdvanced graphical parameters[default =summary] -
Since the dataMatrix does not contain missing value, the singular value decomposition was used by default; NIPALS can be selected with the
Algorithmargument within theAdvanced computational parameters, -
The default (NA)
Number of predictive componentsfor PCA means that components (up to 10) will be computed until the cumulative variance exceeds 50%. If the 50% have not been reached at the 10th component, a warning message will be issued (you can still compute the following components by specifying theNumber of predictive componentsvalue). -
The values of the scores (respectively the loadings) along the two selected components have been added to the [4. Multivariate_sampleMetadata.tsv] (respectively to the [5. Multivariate_variableMetadata.tsv]) tables
-
The values of the variance captured by the 9 components can be seen in the [7. Multivariate_information.txt] text file

For PLS (and OPLS), the Y response(s) must be provided to the opls
method. Y can be either a numeric vector (respectively matrix) for single
(respectively multiple) (O)PLS regression, or a character factor for
(O)PLS-DA classification as in the following example with the gender
qualitative response:
 OPLS-DA model of the gender response. Top left:
OPLS-DA model of the gender response. Top left:
significance diagnostic: the R2Y and Q2Y of the model are compared
with the corresponding values obtained after random permutation of the y
response; Top right: inertia barplot: the graphic here suggests that 3
orthogonal components may be sufficient to capture most of the inertia; Bottom
left: outlier diagnostics; Bottom right: x-score plot: the number of
components and the cumulative R2X, R2Y and Q2Y are indicated below the
plot.
To perform OPLS(-DA), we set orthoI (number of components which are
orthogonal; Integer) to either a specific number of orthogonal components,
or to NA. Let us build an OPLS-DA model of the gender response.
Note:
- When set to NA (as in the default), the number of components
predIis determined automatically as follows [@Eriksson2001]: A new component h is added to the model if:
-
$R2Y_h \geq 0.01$ , i.e., the percentage of Y dispersion (i.e., sum of squares) explained by component h is more than 1 percent, and -
$Q2Y_h=1-PRESS_h/RSS_{h-1} \geq 0$ for PLS (or 5% when the number of samples is less than 100) or 1% for OPLS:$Q2Y_h \geq 0$ means that the predicted residual sum of squares ($PRESS_h$ ) of the model including the new component h estimated by 7-fold cross-validation is less than the residual sum of squares ($RSS_{h-1}$ ) of the model with the previous components only (with$RSS_0$ being the sum of squared Y values).
-
The predictive performance of the full model is assessed by the cumulative Q2Y metric:
$Q2Y=1-\prod\limits_{h=1}^r (1-Q2Y_h)$ . We have$Q2Y \in [0,1]$ , and the higher the Q2Y, the better the performance. Models trained on datasets with a larger number of features compared with the number of samples can be prone to overfitting: in that case, high Q2Y values are obtained by chance only. To estimate the significance of Q2Y (and R2Y) for single response models, permutation testing [@Szymanska2012] can be used: models are built after random permutation of the Y values, and$Q2Y_{perm}$ are computed. The p-value is equal to the proportion of$Q2Y_{perm}$ above$Q2Y$ (the default number of permutations is 20 as a compromise between quality control and computation speed; it can be increased with thepermIparameter, e.g. to 1,000, to assess if the model is significant at the 0.05 level), -
The NIPALS algorithm is used for PLS (and OPLS); dataMatrix matrices with (a moderate amount of) missing values can thus be analysed.
-
For OPLS modeling of a single response, the number of predictive component is 1,
-
In the (
x-scoreplot), the predictive component is displayed as abscissa and the (selected; default = 1) orthogonal component as ordinate.
We see that our model with 3 predictive (pre) components has significant and quite high R2Y and Q2Y values.
The biosigner module, based on the biosigner R/Bioconductor package, implements an innovative method to select variables which significantly contribute to the prediction performance of a binary classifier (i.e. for the discrimination between two classes; Rinaudo et al., 2016): a group of variables is significant if the prediction performance of the classifier decreases when the values of these variables are permutated in a test subset.
The procedure is repeated iteratively by applying the algorithm to the dataset restricted to the significant variables until, for a given round, all candidate variables are found significant (in this case the signature consists of these variables), or until there is no variable left to be tested (in this case the signature is empty). The biosigner module therefore returns a stable final S signature (possibly empty), in addition to several tiers (from A to E) corresponding to the variables discarded during one of the previous iterations (e.g., variables from the A tier were selected in all but the last iteration).
Because test subsets are generated by a random process (bootstrap), the module may generate slightly distinct signatures when re-applied to the same input data. Fixing the Seed in the Advanced computational parameters ensures that results will be identical.
Signatures from three distinct classifiers can be run in parallel, namely Partial Least Square Discriminant Analysis (PLS-DA), Random Forest (RF) and Support Vector Machines (SVM), as the performances of each machine learning approach may vary depending on the structure of the dataset. The signature for each of the selected classifier(s) as an additional column of the variableMetadata table ([15. Biosigner_Multivariate_Univariate_Multivariate_variableMetadata.tsv]). In addition the tiers and individual boxplots of the selected features are plotted (see below).
The module has been successfully applied to transcriptomics and metabolomics data (Rinaudo et al., 2016).
Notes:
- only binary classification is currently available
- if the dataMatrix contains missing values (NA), these features will be removed prior to modeling with Random Forest and SVM (in contrast, the NIPALS algorithm from PLS-DA can handle missing values)
Signatures consisting of 2 (Random Forest) or 3 (PLS-DA, SVM) metabolites (i.e., less than 3% of the initial features) were identified by biosigner ([16: Biosigner__figure-tier.pdf]). Testosterone glucuronide was common to all 3 signatures, and oxoglutaric acid and p-anisic acid to 2 of them. All selected metabolites had a clearcut difference of intensities between males and females ([17: Biosigner__figure-boxplot.pdf]). Prediction accuracies of the models restricted to the signatures (3% of the initial variables) were almost as good as (and superior to for PLS-DA) the models trained on the full dataset ([18: Biosigner__information.txt]).
 Selected variables from the dataset for the PLS-DA, Random Forest, or SVM prediction of gender. The S tier corresponds to the final metabolite signature, i.e., metabolites which passed through all the selection steps.
Selected variables from the dataset for the PLS-DA, Random Forest, or SVM prediction of gender. The S tier corresponds to the final metabolite signature, i.e., metabolites which passed through all the selection steps.
Thank you for working your way through the tutorial and enjoy further analyses with your own data. Feel free to report any feedback to us by email or via the Online Feedback Form. Other tutorials are available on our website (see the PhenoMeNal wiki).
All of these Galaxy modules have been containerized by Pierrick Roger and integrated into the PhenoMeNal instance with the help of Pablo Moreno. Michael van Vliet did a huge work in improving and testing this tutorial.
| Funded by the EC Horizon 2020 programme, grant agreement number 654241 |
|---|
