diff --git a/README.md b/README.md
index 7503685..17b0796 100644
--- a/README.md
+++ b/README.md
@@ -1,91 +1,306 @@
# BismarkPlot
-A small library to plot Bismark ``methylation_extractor`` reports.
+Comprehensive tool for visualizing genome-wide cytosine data.
See the docs: https://shitohana.github.io/BismarkPlot
-Right now only ``coverage2cytosine`` input is supported, but support for ``bismark2bedGraph`` will be added soon.
+Right now only ``coverage2cytosine`` input is supported, but support for other input types will be added soon.
-## Installation
+# Installation
```commandline
pip install bismarkplot
```
-## Usage
+# Console usage
You can use ```bismarkplot``` either as python library or directly from console after installing it.
-To see options:
+
+Console options:
+- *bismarkplot-metagene* - methylation density visualizing tool.
+- *bismarkplot-chrs* - chromosome methylation levels visualizing tool.
+
+### bismarkplot-metagene
+
+```commandline
+usage: BismarkPlot. [-h] [-o OUT] [-g GENOME] [-r {gene,exon,tss,tes}] [-b BATCH] [-c CORES] [-f FLENGTH] [-u UWINDOWS] [-d DWINDOWS] [-m MLENGTH] [-w GWINDOWS] [--line] [--heatmap] [--box] [--violin]
+ [-S SMOOTH] [-L LABELS [LABELS ...]] [-H H] [-V V] [--dpi DPI] [-F {png,pdf,svg}]
+ filename [filename ...]
+
+Metagene visualizing tool.
+
+positional arguments:
+ filename path to bismark methylation_extractor files
+
+options:
+ -h, --help show this help message and exit
+ -o OUT, --out OUT output base name (default: current/path)
+ -g GENOME, --genome GENOME
+ path to GFF genome file (default: None)
+ -r {gene,exon,tss,tes}, --region {gene,exon,tss,tes}
+ path to GFF genome file (default: gene)
+ -b BATCH, --batch BATCH
+ number of rows to be read from bismark file by batch (default: 1000000)
+ -c CORES, --cores CORES
+ number of cores to use (default: None)
+ -f FLENGTH, --flength FLENGTH
+ length in bp of flank regions (default: 2000)
+ -u UWINDOWS, --uwindows UWINDOWS
+ number of windows for upstream (default: 50)
+ -d DWINDOWS, --dwindows DWINDOWS
+ number of windows for downstream (default: 50)
+ -m MLENGTH, --mlength MLENGTH
+ minimal length in bp of gene (default: 4000)
+ -w GWINDOWS, --gwindows GWINDOWS
+ number of windows for genes (default: 100)
+ --line line-plot enabled (default: False)
+ --heatmap heat-map enabled (default: False)
+ --box box-plot enabled (default: False)
+ --violin violin-plot enabled (default: False)
+ -S SMOOTH, --smooth SMOOTH
+ windows for smoothing (default: 10)
+ -L LABELS [LABELS ...], --labels LABELS [LABELS ...]
+ labels for plots (default: None)
+ -H H vertical resolution for heat-map (default: 100)
+ -V V vertical resolution for heat-map (default: 100)
+ --dpi DPI dpi of output plot (default: 200)
+ -F {png,pdf,svg}, --format {png,pdf,svg}
+ format of output plots (default: pdf)
+```
+
+Example:
+
```commandline
-bismarkplot --help
+bismarkplot-metagene -g path/to/genome.gff -r gene -f 2000 -m 4000 -u 500 -d 500 -w 1000 -b 1000000 --line --heatmap --box --violin --dpi 200 -f pdf -S 50 report1.txt report2.txt report3.txt report4.txt
```
-## Examples
-### Example in command line
+[Result](#multiple-samples-same-specie)
+
+### bismarkplot-chrs
+
```commandline
-bismarkplot --genome path/to/genome.gff --flength 2000 --mlength 4000 --fwindows 500 --gwindows 2000 --line-plot --heat-map --box-plot --bar-plot --labels exp1 exp2 --resolution 100 --format pdf path/to/genomeWide1.txt path/to/genomeWide2.txt
+usage: BismarkPlot [-h] [-o DIR] [-b N] [-c CORES] [-w N] [-m N] [-S FLOAT] [-F {png,pdf,svg}] path/to/txt [path/to/txt ...]
+
+Chromosome methylation levels visualization.
+
+positional arguments:
+ path/to/txt path to bismark methylation_extractor file
+
+options:
+ -h, --help show this help message and exit
+ -o DIR, --out DIR output base name (default: current/path)
+ -b N, --batch N number of rows to be read from bismark file by batch (default: 1000000)
+ -c CORES, --cores CORES
+ number of cores to use (default: None)
+ -w N, --wlength N number of windows for genes (default: 100000)
+ -m N, --mlength N minimum chromosome length (default: 1000000)
+ -S FLOAT, --smooth FLOAT
+ windows for smoothing (0 - no smoothing, 1 - straight line (default: 50)
+ -F {png,pdf,svg}, --format {png,pdf,svg}
+ format of output plots (default: pdf)
```
-This command will generate line plots and heat maps pdf files for all contexts and box and bar plots.
-### Example in python script
-If using bismarkplot as python library is more suitable for you, you can call ```bismarkplot``` methods right from python script.
+Example:
+
+```commandline
+bismarkplot-chrs -b 10000000 -w 10000 -m 1000000 -s 10 -f pdf path/to/CX_report.txt
+```
+
+[Result](#chromosome-levels)
+
+# Python
+
+BismarkPlot provides a large variety of function for manipulating with cytosine methylation data.
+
+## Metagene
+
+Below we will show the basic BismarkPlot workflow.
+
+### Single sample
-First we need to initialize ``genome`` and ``BismarkFiles``. ``genome`` is .gff or .bed file with gene coordinates. ``BismarkFiles`` is a class, which calculates data for all plots, so their types need to be specified when it is initialized.
```python
import bismarkplot
+# Firstly, we need to read the regions annotation (e.g. reference genome .gff)
+genome = bismarkplot.Genome.from_gff("path/to/genome.gff")
+# Next we need to filter regions of interest from the genome
+genes = genome.gene_body(min_length=4000, flank_length=2000)
-file = 'path/to/genome.gff'
-
-genome = bismarkplot.read_genome(
- file,
- flank_length=2000,
- min_length=4000
+# Now we need to calculate metagene data
+metagene = bismarkplot.Metagene.from_file(
+ file = "path/to/CX_report.txt",
+ genome=genes, # filtered regions
+ upstream_windows = 500,
+ gene_windows = 1000,
+ downstream_windows = 500,
+ batch_size= 10**7 # number of lines to be read simultaneously
)
-files = ['path/to/genomeWide1.txt', 'path/to/genomeWide2.txt']
-bismark = bismarkplot.BismarkFiles(
- files, genome,
- flank_windows=500,
- gene_windows=2000,
- line_plot=True,
- heat_map=True,
- box_plot=True,
- bar_plot=True
-)
+# Our metagene contains all methylation contexts and both strands, so we need to filter it (as in dplyr)
+filtered = metagene.filter(context = "CG", strand = "+")
+# We are ready to plot
+lp = filtered.line_plot() # line plot data
+lp.draw().savefig("path/to/lp.pdf") # matplotlib.Figure
+
+hm = filtered.heat_map(ncol=200, nrow=200)
+hm.draw().savefig("path/to/hm.pdf") # matplotlib.Figure
```
+Output:
+
+
+  +
+  +
+
-Let's now draw plots themselves.
+### Smoothing the line plot
+
+Smoothing is very useful, when input signal is very weak (e.g. mammalian non-CpG contexts)
-For line plots use (or ``draw_line_plots_all`` for all contexts)
```python
-bismark.draw_line_plots_filtered(
- context='CG',
- strand='+',
- smooth=.05,
- labels = ['exp1', 'exp2'],
- title = 'Plot for CG+'
-)
+# mouse CHG methylation example
+filtered = metagene.filter(context = "CHG", strand = "+")
+lp.draw(smooth = 0).savefig("path/to/lp.pdf") # no smooth
+lp.draw(smooth = 50).savefig("path/to/lp.pdf") # smoothed with window length = 50
```
- +Output:
+
+
+Output:
+
+
+  +
+  +
+
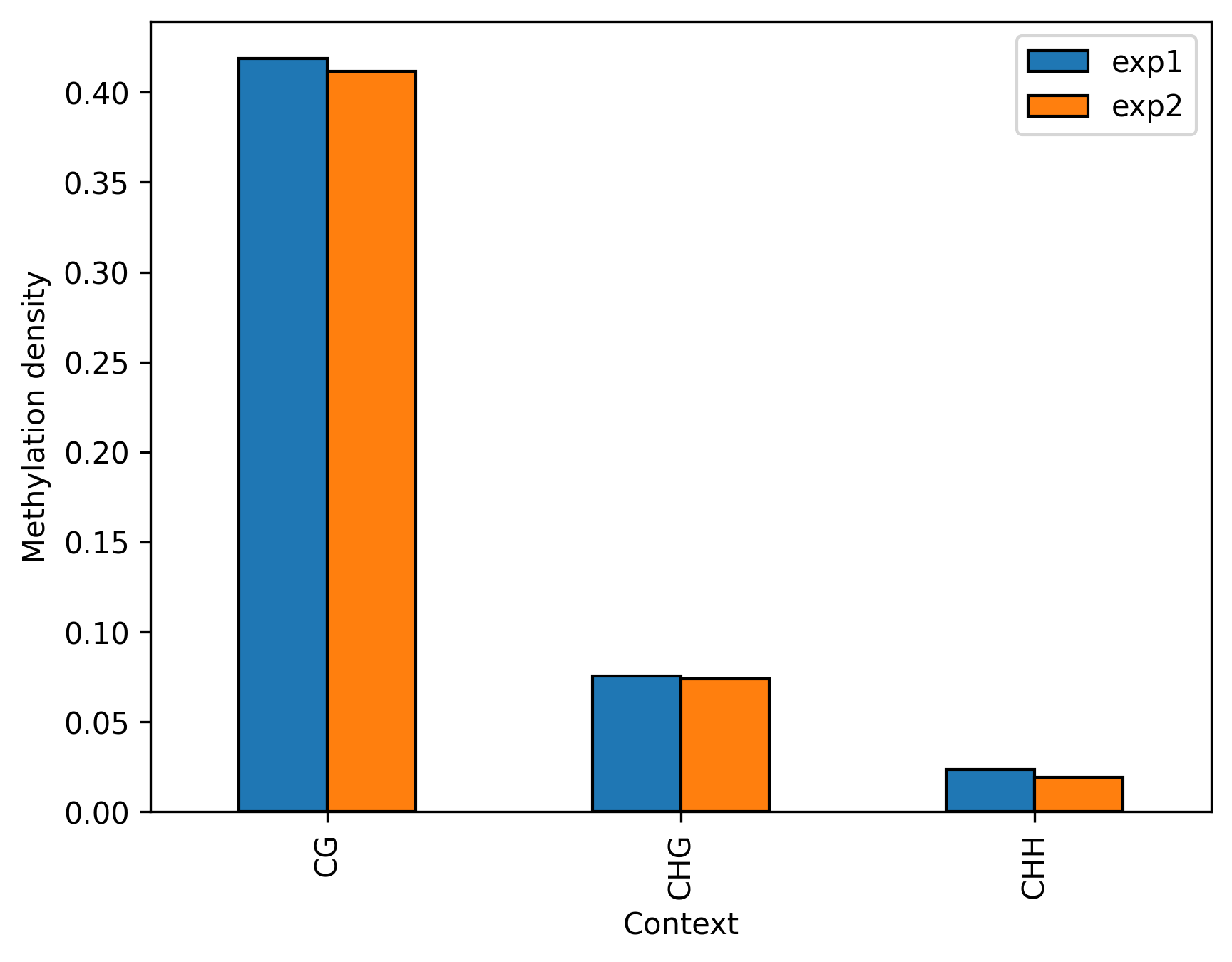
+### Multiple samples, same specie
-For heat maps use (or ``draw_heat_maps_all`` for all contexts)
```python
-bismark.draw_heat_maps_filtered(
- context='CG',
- strand='+',
- resolution=100,
- labels = ['exp1', 'exp2'],
- title = 'Heatmap for CG+'
-)
+# We can initialize genome like in previous example
+
+filenames = ["report1.txt", "report2.txt", "report3.txt", "report4.txt"]
+metagenes = bismarkplot.MetageneFiles.from_list(filenames, labels = ["1", "2", "3", "4"], ...) # rest of params from previous example
+
+# Our metagenes contains all methylation contexts and both strands, so we need to filter it (as in dplyr)
+filtered = metagenes.filter(context = "CG", strand = "+")
+
+# Now we can draw line-plot or heatmap like in previous example, or plot distribution statistics as shown below
+trimmed = filtered.trim_flank() # we want to analyze only gene bodies
+trimmed.box_plot(showfliers=False).savefig(...)
+trimmed.violin_plot().savefig(...)
+
+# If data is technical replicates we can merge them into single DataFrame and analyze as one
+merged = filtered.merge()
+```
+
+Output:
+
+
+  +
+  +
+
+
+  +
+  +
+
+
+### Multiple samples, multiple species
+
+```python
+# For analyzing samples with different reference genomes, we need to initialize several genomes instances
+genome_filenames = ["arabidopsis.gff", "brachypodium.gff", "cucumis.gff", "mus.gff"]
+reports_filenames = ["arabidopsis.txt", "brachypodium.txt", "cucumis.txt", "mus.txt"]
+
+genomes = [
+ bismarkplot.Genome.from_gff(file).gene_body(...) for file in genome_filenames
+]
+
+# Now we read reports
+metagenes = []
+for report, genome in zip(reports_filenames, genomes):
+ metagene = bismarkplot.Metagene(report, genome = genome, ...)
+ metagenes.append(metagene)
+
+# Initialize MetageneFiles
+labels = ["A. thaliana", "B. distachyon", "C. sativus", "M. musculus"]
+metagenes = Bismarkplot.MetageneFiles(metagenes, labels)
+# Now we can plot them like in previous example
+```
+
+Output:
+
+
+  +
+  +
+
+
+  +
+  +
+
+
+### Different regions
+
+Other genomic regions from .gff can be analyzed too with ```.exon``` or ```.near_tss/.near_tes``` option for ```bismarkplot.Genome```
+
+```python
+exons = [
+ bismarkplot.Genome.from_gff(file).exon(min_length=100) for file in genome_filenames
+]
+metagenes = []
+for report, exon in zip(reports_filenames, exons):
+ metagene = bismarkplot.Metagene(report, genome = exon,
+ upstream_windows = 0, # !!!
+ downstream_windows = 0, # !!!
+ ...)
+ metagenes.append(metagene)
+# OR
+tss = [
+ bismarkplot.Genome.from_gff(file).near_tss(min_length = 2000, flank_length = 2000) for file in genome_filenames
+]
+metagenes = []
+for report, t in zip(reports_filenames, tss):
+ metagene = bismarkplot.Metagene(report, genome = t,
+ upstream_windows = 1000,# same number of windows
+ gene_windows = 1000, # same number of windows
+ downstream_windows = 0, # !!!
+ ...)
+ metagenes.append(metagene)
```
- +Exon output:
+
+Exon output:
+
+  +
+  +
+
+
+TSS output:
+
+  +
+
+
+## Chromosome levels
+
+BismarkPlot allows user to visualize chromosome methylation levels across full genome
-For box plot or bar plot use
```python
-bismark.draw_box_plot(strand_specific=True, labels=['exp1', 'exp2'])
-bismark.draw_bar_plot(labels=['exp1', 'exp2'])
+import bismarkplot
+chr = bismarkplot.ChrLevels.from_file(
+ "path/to/CX_report.txt",
+ window_length=10**5, # window length in bp
+ batch_size=10**7,
+ chr_min_length = 10**6, # minimum chr length in bp
+)
+fig, axes = plt.subplots()
+
+for context in ["CG", "CHG", "CHH"]:
+ chr.filter(strand="+", context=context).draw(
+ (fig, axes), # to plot contexts on same axes
+ smooth=10, # window number for smoothing
+ label=context # labels for lines
+ )
+
+fig.savefig(f"chrom.pdf", dpi = 200)
```
- -
- +
+Output for Arabidopsis t.:
+
+
+
+Output for Arabidopsis t.:
+
+ +
+Output for Brachypodium d.:
+
+
+
+Output for Brachypodium d.:
+
+ \ No newline at end of file
diff --git a/src/bismarkplot/console_chrs.py b/src/bismarkplot/console_chrs.py
index 893d528..d7752cc 100644
--- a/src/bismarkplot/console_chrs.py
+++ b/src/bismarkplot/console_chrs.py
@@ -5,7 +5,8 @@
parser = argparse.ArgumentParser(
prog='BismarkPlot',
- description='Chromosome methylation levels visualization.'
+ description='Chromosome methylation levels visualization.',
+ formatter_class=argparse.ArgumentDefaultsHelpFormatter
)
parser.add_argument('filename', help='path to bismark methylation_extractor file', nargs='+', metavar='path/to/txt')
@@ -46,3 +47,7 @@ def main():
with open(args.out + '/' + filename, 'w') as f:
f.write(traceback.format_exc())
print(f'Error happened. Please open an issue at GitHub with Traceback from file: {f}')
+
+
+if __name__ == "__main__":
+ main()
\ No newline at end of file
diff --git a/src/bismarkplot/console_metagene.py b/src/bismarkplot/console_metagene.py
index 1fd2611..e1cd26d 100644
--- a/src/bismarkplot/console_metagene.py
+++ b/src/bismarkplot/console_metagene.py
@@ -5,31 +5,31 @@
parser = argparse.ArgumentParser(
prog='BismarkPlot.',
- description='Metagene visualizing tool.'
+ description='Metagene visualizing tool.',
+ formatter_class=argparse.ArgumentDefaultsHelpFormatter
)
-
-parser.add_argument('filename', help='path to bismark methylation_extractor files', nargs='+', metavar='path/to/txt')
-parser.add_argument('-o', '--out', help='output base name', default=os.path.abspath(os.getcwd()), metavar='DIR')
-parser.add_argument('-g', '--genome', help='path to GFF genome file', metavar='/path/to/gff')
-parser.add_argument('-r', '--region', help='path to GFF genome file', metavar='/path/to/gff', default = "gene", choices=["gene", "exon", "tss", "tes"])
-parser.add_argument('-b', '--batch', help='number of rows to be read from bismark file by batch', type=int, default=10**6, metavar='N')
+parser.add_argument('filename', help='path to bismark methylation_extractor files', nargs='+')
+parser.add_argument('-o', '--out', help='output base name', default=os.path.abspath(os.getcwd()))
+parser.add_argument('-g', '--genome', help='path to GFF genome file')
+parser.add_argument('-r', '--region', help='path to GFF genome file', default = "gene", choices=["gene", "exon", "tss", "tes"])
+parser.add_argument('-b', '--batch', help='number of rows to be read from bismark file by batch', type=int, default=10**6)
parser.add_argument('-c', '--cores', help='number of cores to use', type=int, default=None)
-parser.add_argument('-f', '--flength', help='length in bp of flank regions', type=int, default=2000, metavar='LENGTH')
-parser.add_argument('-u', '--uwindows', help='number of windows for upstream', type=int, default=50, metavar='N')
-parser.add_argument('-d', '--dwindows', help='number of windows for downstream', type=int, default=50, metavar='N')
-parser.add_argument('-m', '--mlength', help='minimal length in bp of gene', type=int, default=4000, metavar='LENGTH')
-parser.add_argument('-w', '--gwindows', help='number of windows for genes', type=int, default=100, metavar='N')
+parser.add_argument('-f', '--flength', help='length in bp of flank regions', type=int, default=2000)
+parser.add_argument('-u', '--uwindows', help='number of windows for upstream', type=int, default=50)
+parser.add_argument('-d', '--dwindows', help='number of windows for downstream', type=int, default=50)
+parser.add_argument('-m', '--mlength', help='minimal length in bp of gene', type=int, default=4000)
+parser.add_argument('-w', '--gwindows', help='number of windows for genes', type=int, default=100)
-parser.add_argument('-LP', '--line-plot', help='line-plot enabled', action='store_true')
-parser.add_argument('-HM', '--heat-map', help='heat-map enabled', action='store_true')
-parser.add_argument('-BX', '--box-plot', help='box-plot enabled', action='store_true')
-parser.add_argument('-VL', '--violin-plot', help='bar-plot enabled', action='store_true')
+parser.add_argument('--line', help='line-plot enabled', action='store_true')
+parser.add_argument('--heatmap', help='heat-map enabled', action='store_true')
+parser.add_argument('--box', help='box-plot enabled', action='store_true')
+parser.add_argument('--violin', help='violin-plot enabled', action='store_true')
-parser.add_argument('-S', '--smooth', help='windows for smoothing (0 - no smoothing, 1 - straight line', type=float, default=10, metavar='FLOAT')
-parser.add_argument('-L', '--labels', help='labels for plots', nargs='+', metavar='NAME')
-parser.add_argument('-HR', '--hresolution', help='vertical resolution for heat-map', type=int, metavar='N', default=100)
-parser.add_argument('-VR', '--vresolution', help='vertical resolution for heat-map', type=int, metavar='N', default=100)
-parser.add_argument("--dpi", help="dpi of output plot", type=int, metavar="N", default=200)
+parser.add_argument('-S', '--smooth', help='windows for smoothing', type=float, default=10)
+parser.add_argument('-L', '--labels', help='labels for plots', nargs='+')
+parser.add_argument('-H', help='vertical resolution for heat-map', type=int, default=100)
+parser.add_argument('-V', help='vertical resolution for heat-map', type=int, default=100)
+parser.add_argument("--dpi", help="dpi of output plot", type=int, default=200)
parser.add_argument('-F', '--format', help='format of output plots', choices=['png', 'pdf', 'svg'], default='pdf', dest='file_format')
@@ -82,3 +82,7 @@ def main():
with open(args.out + '/' + filename, 'w') as f:
f.write(traceback.format_exc())
print(f'Error happened. Please open an issue at GitHub with Traceback from file: {f}')
+
+
+if __name__ == "__main__":
+ main()
\ No newline at end of file
\ No newline at end of file
diff --git a/src/bismarkplot/console_chrs.py b/src/bismarkplot/console_chrs.py
index 893d528..d7752cc 100644
--- a/src/bismarkplot/console_chrs.py
+++ b/src/bismarkplot/console_chrs.py
@@ -5,7 +5,8 @@
parser = argparse.ArgumentParser(
prog='BismarkPlot',
- description='Chromosome methylation levels visualization.'
+ description='Chromosome methylation levels visualization.',
+ formatter_class=argparse.ArgumentDefaultsHelpFormatter
)
parser.add_argument('filename', help='path to bismark methylation_extractor file', nargs='+', metavar='path/to/txt')
@@ -46,3 +47,7 @@ def main():
with open(args.out + '/' + filename, 'w') as f:
f.write(traceback.format_exc())
print(f'Error happened. Please open an issue at GitHub with Traceback from file: {f}')
+
+
+if __name__ == "__main__":
+ main()
\ No newline at end of file
diff --git a/src/bismarkplot/console_metagene.py b/src/bismarkplot/console_metagene.py
index 1fd2611..e1cd26d 100644
--- a/src/bismarkplot/console_metagene.py
+++ b/src/bismarkplot/console_metagene.py
@@ -5,31 +5,31 @@
parser = argparse.ArgumentParser(
prog='BismarkPlot.',
- description='Metagene visualizing tool.'
+ description='Metagene visualizing tool.',
+ formatter_class=argparse.ArgumentDefaultsHelpFormatter
)
-
-parser.add_argument('filename', help='path to bismark methylation_extractor files', nargs='+', metavar='path/to/txt')
-parser.add_argument('-o', '--out', help='output base name', default=os.path.abspath(os.getcwd()), metavar='DIR')
-parser.add_argument('-g', '--genome', help='path to GFF genome file', metavar='/path/to/gff')
-parser.add_argument('-r', '--region', help='path to GFF genome file', metavar='/path/to/gff', default = "gene", choices=["gene", "exon", "tss", "tes"])
-parser.add_argument('-b', '--batch', help='number of rows to be read from bismark file by batch', type=int, default=10**6, metavar='N')
+parser.add_argument('filename', help='path to bismark methylation_extractor files', nargs='+')
+parser.add_argument('-o', '--out', help='output base name', default=os.path.abspath(os.getcwd()))
+parser.add_argument('-g', '--genome', help='path to GFF genome file')
+parser.add_argument('-r', '--region', help='path to GFF genome file', default = "gene", choices=["gene", "exon", "tss", "tes"])
+parser.add_argument('-b', '--batch', help='number of rows to be read from bismark file by batch', type=int, default=10**6)
parser.add_argument('-c', '--cores', help='number of cores to use', type=int, default=None)
-parser.add_argument('-f', '--flength', help='length in bp of flank regions', type=int, default=2000, metavar='LENGTH')
-parser.add_argument('-u', '--uwindows', help='number of windows for upstream', type=int, default=50, metavar='N')
-parser.add_argument('-d', '--dwindows', help='number of windows for downstream', type=int, default=50, metavar='N')
-parser.add_argument('-m', '--mlength', help='minimal length in bp of gene', type=int, default=4000, metavar='LENGTH')
-parser.add_argument('-w', '--gwindows', help='number of windows for genes', type=int, default=100, metavar='N')
+parser.add_argument('-f', '--flength', help='length in bp of flank regions', type=int, default=2000)
+parser.add_argument('-u', '--uwindows', help='number of windows for upstream', type=int, default=50)
+parser.add_argument('-d', '--dwindows', help='number of windows for downstream', type=int, default=50)
+parser.add_argument('-m', '--mlength', help='minimal length in bp of gene', type=int, default=4000)
+parser.add_argument('-w', '--gwindows', help='number of windows for genes', type=int, default=100)
-parser.add_argument('-LP', '--line-plot', help='line-plot enabled', action='store_true')
-parser.add_argument('-HM', '--heat-map', help='heat-map enabled', action='store_true')
-parser.add_argument('-BX', '--box-plot', help='box-plot enabled', action='store_true')
-parser.add_argument('-VL', '--violin-plot', help='bar-plot enabled', action='store_true')
+parser.add_argument('--line', help='line-plot enabled', action='store_true')
+parser.add_argument('--heatmap', help='heat-map enabled', action='store_true')
+parser.add_argument('--box', help='box-plot enabled', action='store_true')
+parser.add_argument('--violin', help='violin-plot enabled', action='store_true')
-parser.add_argument('-S', '--smooth', help='windows for smoothing (0 - no smoothing, 1 - straight line', type=float, default=10, metavar='FLOAT')
-parser.add_argument('-L', '--labels', help='labels for plots', nargs='+', metavar='NAME')
-parser.add_argument('-HR', '--hresolution', help='vertical resolution for heat-map', type=int, metavar='N', default=100)
-parser.add_argument('-VR', '--vresolution', help='vertical resolution for heat-map', type=int, metavar='N', default=100)
-parser.add_argument("--dpi", help="dpi of output plot", type=int, metavar="N", default=200)
+parser.add_argument('-S', '--smooth', help='windows for smoothing', type=float, default=10)
+parser.add_argument('-L', '--labels', help='labels for plots', nargs='+')
+parser.add_argument('-H', help='vertical resolution for heat-map', type=int, default=100)
+parser.add_argument('-V', help='vertical resolution for heat-map', type=int, default=100)
+parser.add_argument("--dpi", help="dpi of output plot", type=int, default=200)
parser.add_argument('-F', '--format', help='format of output plots', choices=['png', 'pdf', 'svg'], default='pdf', dest='file_format')
@@ -82,3 +82,7 @@ def main():
with open(args.out + '/' + filename, 'w') as f:
f.write(traceback.format_exc())
print(f'Error happened. Please open an issue at GitHub with Traceback from file: {f}')
+
+
+if __name__ == "__main__":
+ main()
\ No newline at end of file