Tutorial
This tutorial provide a step-by-step analysis using the PanPhlAn software to map a samples against the E.rectale species pangenome.
- 1. Download and install PanPhlAn
- 2. Download the metagenomics samples
- 3. Download the reference genomes
- 4. Build bowtie2 indexes
- 5. Map samples against pangenome
- 6. Profiling strains
- Visualizing the matrix
git clone https://github.com/SegataLab/panphlan.git
see also Download and Installation for further details
In this example, we use 25 samples coming from various datasets (different studies, pathologies, country, westernized and non-westernized...). Sample can be downloaded using the following command :
wget https://www.dropbox.com/s/oi26jg0v7ktlavc/panphlan_tutorial_samples.tar.bz2
Then, the archive must be extracted in order to get one file per sample
tar -xvjf panphlan_tutorial_samples.tar.bz2
25 fastq files are now available in a samples_fastq/ folder. They correspond to various metagenomes samples from datasets spanning across studies and countries.
sampleID Dataset Country
CCMD34381688ST-21-0 ZellerG_2014 DEU
G78505 VatanenT_2016 RUS
G88884 SchirmerM_2016 NLD
G88970 SchirmerM_2016 NLD
G89027 SchirmerM_2016 NLD
H2M514903 LiJ_2017 CHN
H3M518116 LiJ_2017 CHN
HD-1 QinN_2014 CHN
HD-5 QinN_2014 CHN
HV-6 QinN_2014 CHN
LD-48 QinN_2014 CHN
M1.48.ST BritoIL_2016 FJI
M2.48.ST BritoIL_2016 REF
M2.58.ST BritoIL_2016 FJI
N021 WenC_2017 CHN
RA023 WenC_2017 CHN
S353 KarlssonFH_2013 SWE
SID530054 FengQ_2015 AUT
SRS011302 HMP_2012 USA
SZAXPI003417-4 YuJ_2015 CHN
T2D-025 QinJ_2012 CHN
T2D-063 QinJ_2012 CHN
T2D-105 QinJ_2012 CHN
W1.27.ST BritoIL_2016 FJI
YSZC12003_36795 XieH_2016 GBR
For E.rectale (NCBI taxonomy ID : 39491), 15 reference genomes are available from NCBI assembly
GCA_000209935.fna
GCA_000209955.fna
GCA_001404855.fna
GCA_001405295.fna
GCA_001406375.fna
GCA_001406835.fna
GCA_003122495.fna
GCA_003436035.fna
GCA_003436785.fna
GCA_003438175.fna
GCA_003438365.fna
GCA_003438715.fna
GCA_003438925.fna
GCA_003438965.fna
Let's store them in a folder called reference_genomes/.
One also need the pangenome file panphlan_39491_pangenome.csv from CHocoPlhAn export countaining information about gene families UniRef90 ID, gene family name and position (genome, contig, start and stop)
head panphlan_erectale_pangenome.csv
UniRef90_A0A395V045 EUBREC_3309 GCA_000020605 CP001107.1 3142535 3142891
UniRef90_C4ZFP9 EUBREC_0863 GCA_000020605 CP001107.1 771233 772936
UniRef90_C4Z809 EUBREC_1266 GCA_000020605 CP001107.1 1157854 1158015
UniRef90_A0A174M4W1 EUBREC_2028 GCA_000020605 CP001107.1 1915753 1916655
UniRef90_A0A3E4WQ94 EUBREC_3406 GCA_000020605 CP001107.1 3214586 3216313
UniRef90_A0A395ZCP1 EUBREC_3083 GCA_000020605 CP001107.1 2948412 2949056
UniRef90_A0A0M6WH97 EUBREC_1428 GCA_000020605 CP001107.1 1312977 1313234
UniRef90_C4ZCE9 EUBREC_2154 GCA_000020605 CP001107.1 2006055 2006699
UniRef90_A0A396FK33 EUBREC_0251 GCA_000020605 CP001107.1 233885 235228
- Since the pangenome file is named
panphlan_39491_pangenome.csv, the clade name argument should take the value39491(E.rectale taxonomy ID on NCBI). - Previously downloaded references genomes should be stored in the folder gave as input argument
reference_genomes/. - The output will be generated in the current repository (
-o .) but another folder can be specified.
./panphlan/panphlan_new_pangenome_generation.py -c 39491 --i_fna reference_genomes/ -o . --verbose
Files generated are:
- 6 bowtie2 indexes files named
panphlan_39491.[1-4].bt2andpanphlan_39491.rev.[1-2].bt2 -
39491_ref_genomes.fnacontaining the concatenation of the.fnafiles from the input folder
Here is one example for a sample.
panphlan/panphlan_map.py -c 39491 -i CCMD34381688ST-21-0.fastq.bz2 -o map_results/CCMD34381688ST-21-0_erectale.csv
If the output folder map_results/ does not exist, it will be created.
The clade name has to be specified so that the software can link data from the pangenome file (UniRef90 ID mainly) to the location mapped in the genome.
The script has to be run one time for each sample. If samples are numerous, GNU parallel can be a relevant option.
For a quicker progress through this tutorial, the results from the mapping process for all 25 samples can be directly downloaded :
wget https://www.dropbox.com/s/z81e87dvtzp6pu3/panphlan_tutorial_map_results.tar.bz2
tar -xvjf panphlan_tutorial_map_results.tar.bz2
The most important part of the PanPhlAn software is the profiling part (panphlan_profile.py).
panphlan/panphlan_profile.py -c 39491 -i map_results_erectale/ -o profile_erectale.csv --verbose
This line will generate the presence/absence matrix of gene families. 1 means the gene family is present in the sample, 0 it is not.
CCMD34381688ST-21-0 G78505 G88884 G88970 G89027 H2M514903 H3M518116 HD-1 HD-5 HV-6 LD-48 M1 M2_48 M2_58 N021 RA023 S353 SID530054 SRS011302 SZAXPI003417-4 T2D-025 T2D-063 T2D-105 W1 YSZC12003_36795
UniRef90_A0A069A530 0 0 0 0 0 1 1 1 0 0 0 0 0 0 0 0 0 0 1 0 0 0 1 0 0
UniRef90_A0A076YZQ3 1 0 1 0 0 0 1 0 1 0 1 0 0 0 0 0 0 1 1 0 0 0 1 1 1
UniRef90_A0A095XLP0 1 0 1 1 1 0 0 0 0 0 1 0 0 0 1 1 1 1 1 0 0 0 0 1 1
UniRef90_A0A095XNF2 1 0 1 1 1 0 0 0 0 0 1 0 0 0 1 1 1 1 1 0 0 0 0 1 1
UniRef90_A0A095ZX26 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 1 0 0 0 0 0 1
UniRef90_A0A0D0SCG4 0 0 1 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0
In order to assess whether or not a gene family is present in a sample, PanPlhAn draws a coverage curve of gene families for each sample. Such curve can be obtained using the --o_covplot erectale_covplot creating the erectale_covplot.png figure :

Here, the peak on the leftmost part of the plot has been cut in order to highlight the plateau and the differences between sample. The option --covplot_ymax 100 has been used.
PanPhlAn's aim is to detect a plateau in the coverage curve. Default threshold can be changed for more or less sensitive options. Too stringent option lead to discarding a sample from the matrix as the species will be judged non present in the sample.
Least stringent option is performed by adding --min_coverage 1 --left_max 1.70 --right_min 0.30 in the command line.
By default only the mapped samples whose coverage curves display a satisfying plateau are present in the final matrix. In order to add the reference genomes in the matrix as well, the option --add_strains must be specified.
Moreover, if a specific file mapping UniRef90 gene families names to other annotation data is provided, it can be added in the matrix. For example :
head HUMAnN2_CHOCOPhlAn_201901_functional_annotation_mapping.tsv
NR90 NR50 GO KO KEGG Pfam EC eggNOG
UniRef90_A0A1Y3PFA4 UniRef50_A0A1Y3PFA4 PF13701
UniRef90_A0A1Y3PSN1 UniRef50_A9W6U1 GO:0005623,GO:0016209,GO:0016491,GO:0045454 PF00578
UniRef90_A0A1Y3PG68 UniRef50_L5MXD7 GO:0009228 PF09084
UniRef90_A0A1Y3PRQ8 UniRef50_A0A1Y3PRQ8 PF12172,PF01796
UniRef90_A0A1Y3PNS7 UniRef50_Q1QWU2 GO:0016021 PF14667
UniRef90_A0A1Y3PAH9 UniRef50_A0A0A2VHC2 GO:0016021 PF04239
UniRef90_A0A1Y3PT72 UniRef50_A0A1Y3PT72 GO:0005886,GO:0003755,GO:0006457 5.2.1.8
UniRef90_A0A1Y3PHG5 UniRef50_A0A1L8I4N7 GO:0003677,GO:0003700 PF00027,PF13545
UniRef90_A0A1Y3PFM9 UniRef50_A0A1Y3PFM9 GO:0016021,GO:0005524,GO:0000155 PF02518,PF00512
given as argument as in here:
panphlan/panphlan_profile.py -c 39491 -i map_results/ -o profile_erectale.csv --funct_annot HUMAnN2_CHOCOPhlAn_201901_functional_annotation_mapping.tsv --verbose
will add an annotation column in the presence/absence matrix. Here, without any specification, the second field is taken (UniRef50) but it can be chosen via the --field argument (by default --field has value 1, thus using the second field of the annotation file).
Using the matrix .csv file, visualization can be made through PCoA plots or Heatmaps of gene families presence/absence fingerprint for each sample.
For example, a PCoA of E.rectale strains highlights the existence of westernized (AUT, USA, NLD, SWE, GBR...) cluster compared to a non-westernized (CHN) one.
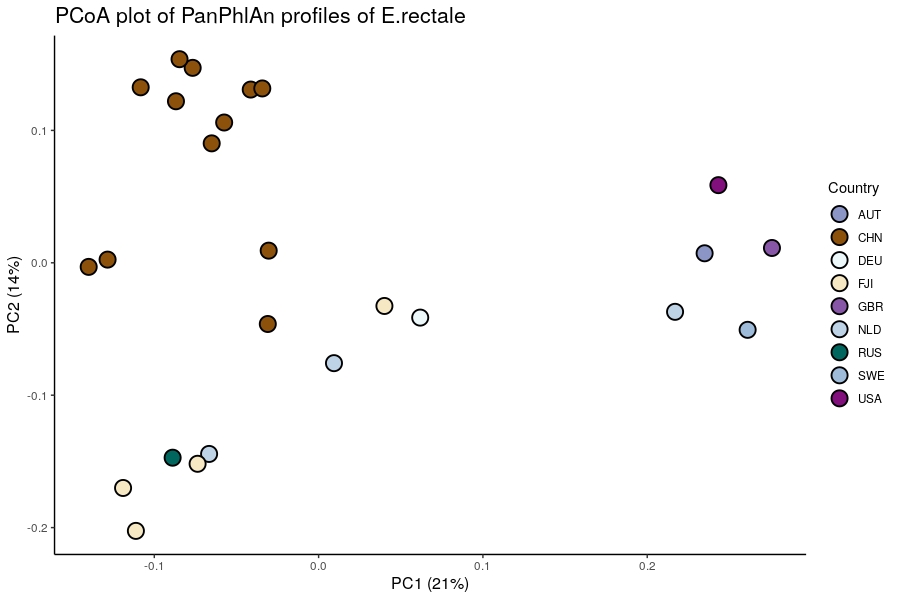
Such clusters can also be visualized through heatmap and hierarchical clustering using the same color code for countries. On such Heatmap, the core genome can also be visualized (yellow columns on the right)

keep same example for R.bromii?