Bioconvert is a collaborative project to facilitate the interconversion of life science data from one format to another.




| contributions: | Want to add a convertor ? Please join #1 |
|---|---|
| How to cite: | Caro et al, BioConvert: a comprehensive format converter for life sciences (2023) NAR Genomics and Bioinformatics (5),3. https://doi.org/10.1093/nargab/lqad074 |
Life science uses many different formats. They may be old, or with complex syntax and converting those formats may be a challenge. Bioconvert aims at providing a common tool / interface to convert life science data formats from one to another.
Many conversion tools already exist but they may be dispersed, focused on few specific formats, difficult to install, or not optimised. With Bioconvert, we plan to cover a wide spectrum of format conversions; we will re-use existing tools when possible and provide facilities to compare different conversion tools or methods via benchmarking. New implementations are provided when considered better than existing ones.
In Jan 2023, we had 50 formats, 100 direct conversions available.
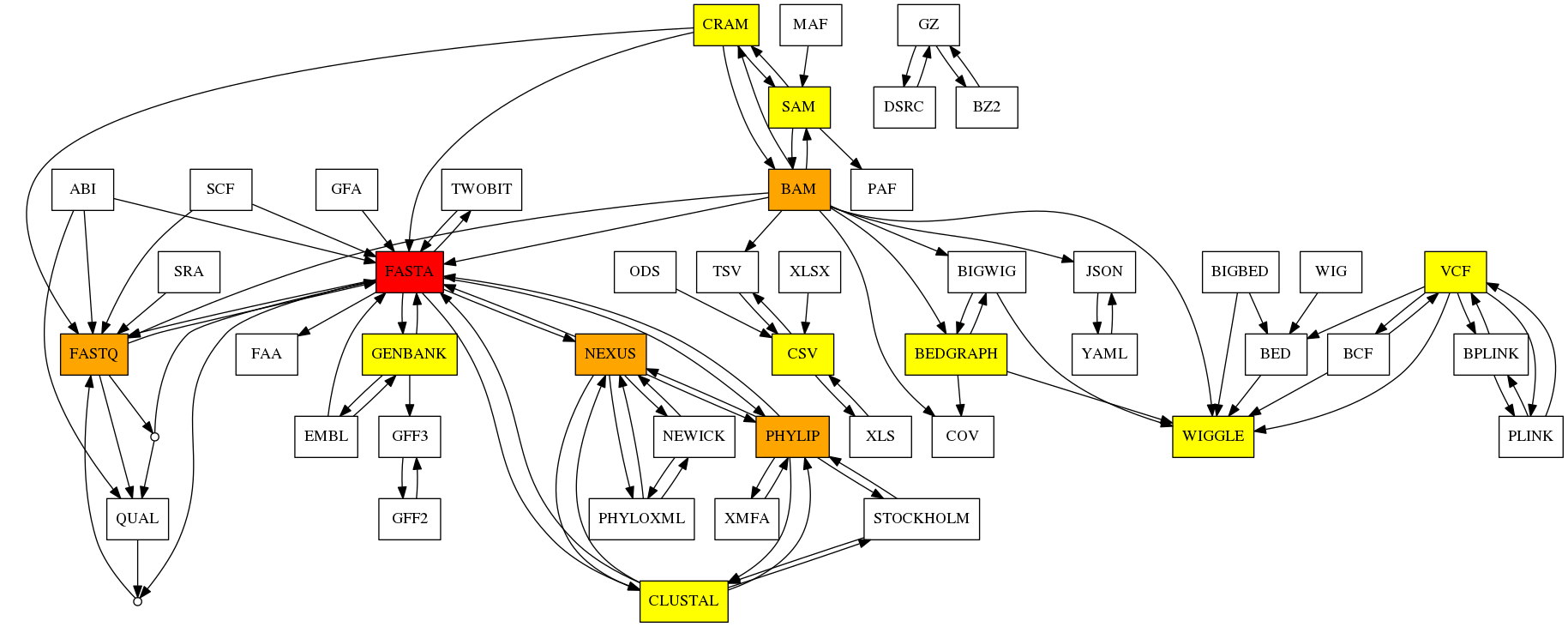
BioConvert is developped in Python. Please use conda or any Python environment manager to install BioConvert using the pip command:
pip install bioconvert
50% of the conversions should work out of the box. However, many conversions require external tools. This is why we recommend to use a conda environment. In particular, most external tools are available on the bioconda channel. For instance if you want to convert a SAM file to a BAM file you would need to install samtools as follow:
conda install -c bioconda samtools
Since bioconvert is available on bioconda on solution that installs BioConvert and all its dependencies is to use conda/mamba:
conda env create --name bioconvert mamba conda activate bioconvert mamba install bioconvert bioconvert --help
See the Installation section for more details and alternative solutions (docker, singularity).
There are many conversions available. Type:
bioconvert --help
to get a list of valid method of conversions. Taking the example of a conversion from a FastQ file into a FastA file, you could do the conversion as follows:
bioconvert fastq2fasta input.fastq output.fasta bioconvert fastq2fasta input.fq output.fasta bioconvert fastq2fasta input.fq.gz output.fasta.gz bioconvert fastq2fasta input.fq.gz output.fasta.bz2
When there is no ambiguity, you can be implicit:
bioconvert input.fastq output.fasta
The default method of conversion is used but you may use another one. Checkout the available methods with:
bioconvert fastq2fasta --show-methods
For more help about a conversion, just type:
bioconvert fastq2fasta --help
and more generally:
bioconvert --help
You may also call BioConvert from a Python shell:
# import a converter from bioconvert.fastq2fasta import FASTQ2FASTA # Instanciate with infile/outfile names convert = FASTQ2FASTA(infile, outfile) # the conversion itself: convert()
{kind=link}
Setting up and maintaining Bioconvert has been possible thanks to users and contributors. Thanks to all:
| Version | Description |
|---|---|
| 1.1.1 |
|
| 1.1.0 |
|
| 1.0.0 |
|
| 0.6.3 |
|
| 0.6.2 |
|
| 0.6.1 |
|
| 0.6.0 |
|
| 0.5.2 |
|
| 0.5.1 |
|
| 0.5.0 |
|
| 0.4.X |
|
| 0.3.X | may 2019. new methods abi2qual, bigbed2bed, etc. added --threads option |
| 0.2.X | aug 2018. abi2fastx, bioconvert_stats tool added |
| 0.1.X | major refactoring to have subcommands with implicit/explicit mode |