Home
Welcome to my bioinformatics top 10 list of tools and tips for molecular microbiology and microbiome software development!
Smith-Waterman local alignment tool that uses a seed/kmer based indexing heuristic to minimize the alignment space. The New York Times calls it the "Google of biological research". It is the most widely used Bioinformatics tool.
https://blast.ncbi.nlm.nih.gov/

https://ftp.ncbi.nlm.nih.gov/blast/executables/LATEST/

https://quay.io/repository/biocontainers/blast

OS-level virtual environments that provides isolation of software, libraries and configurations.
- Isolation of application and package dependencies
- Pipeline reproducibility
bash shortcut:
% cat dr
#!/usr/bin/env bash
pattern=$1
shift 1
image=$(docker images --format "{{.Repository}}:{{.Tag}}" | grep $pattern | head --lines 1)
docker run --volume $(pwd):$(pwd) --workdir $(pwd) $image $@
example usage
% docker images
REPOSITORY TAG IMAGE ID CREATED SIZE
ubuntu 18.04 2c047404e52d 2 weeks ago 63.3MB
quay.io/biocontainers/infernal 1.1.3--h516909a_0 b679b44fb51b 3 months ago 42.5MB
% dr infernal:1.1.3 echo hello world
hello world
Pipeline frameworks and libraries for cloud computing and on-demand scaling of compute resources
- https://www.nextflow.io
- https://github.com/crosenth/scons_batch
- https://github.com/crosenth/aws_batch
% nextflow run tutorial.nf --executor awsbatch --queue my-queue
N E X T F L O W ~ version 19.04.0
executor > my-queue (3)
[69/c8ea4a] process > splitLetters [100%] 1 of 1 ✔
[84/c8b7f1] process > convertToUpper [100%] 2 of 2 ✔
HELLO
WORLD!
% scons -v --executor awsbatch --queue my-queue
SUBMITTED
RUNNABLE
STARTING
printf 'HELLO WORLD!' | split -b 6 - chunk_
SUCCEEDED
SUBMITTED
RUNNABLE
STARTING
cat chunk_aa | tr '[a-z]' '[A-Z]'
cat chunk_ab | tr '[a-z]' '[A-Z]'
HELLO
WORLD!
SUCCEEDED
% aws_batch -v --job-queue my-queue --command "nextflow run tutorial.nf --executor local" quay-io-biocontainers-nextflow
SUBMITTED
RUNNABLE
STARTING
N E X T F L O W ~ version 20.10.0
executor > local (3)
[69/c8ea4a] process > splitLetters [100%] 1 of 1 ✔
[84/c8b7f1] process > convertToUpper [100%] 2 of 2 ✔
HELLO
WORLD!
SUCCEEDED
Command-line tools for converting, modifying and visualizing csv/tsv files
bash shortcut:
% cat cl
#!/usr/bin/env bash
csvpandas look "$@" | less -S
example usage:
% cl blast.csv
|------+---------+----------------------------------+------------|
| name | rank | tax_name | likelihood |
|------+---------+----------------------------------+------------|
| seq1 | species | Streptococcus cristatus | 0.9284 |
| seq2 | species | Streptococcus parasanguinis | 0.9991 |
| seq3 | genus | Streptococcus | 0.9588 |
| seq4 | species | Streptococcus pneumoniae | 0.9806 |
| seq5 | | DISTANT | 0.7237 |
| seq6 | | DISTANT | 1.0 |
| seq7 | genus | Streptococcus | 0.8751 |
| seq8 | species | Streptococcus cristatus | 0.8653 |
|------+---------+----------------------------------+------------|
Python library of bioinformatics tools for pipeline scripting and application development
example usage:
% cat example.py
#!/usr/bin/env python3
import sys
from Bio import SeqIO, Phylo #, ...
if __name__ == '__main__':
for sequence in SeqIO.parse('sequence.fasta', 'fasta'):
print(sequence)
sys.exit()
A multiprocessed utility for retrieving records and parsing feature tables from NCBI
https://github.com/crosenth/medirect
% mefetch -id KN150849 -db nucleotide -email [email protected] -format ft | ftract --feature rrna:product:16s
id,seq_start,seq_stop,strand
KN150849.1,594136,595654,2
KN150849.1,807985,809503,2
KN150849.1,2227751,2229271,1
% mefetch -id accessions.txt -db nucleotide -email [email protected] -format ft -proc 8 | ftract --feature rrna:product:16s | mefetch -db nucleotide -email [email protected] -csv -format gb -out 16s.gb -proc 8
Command-line tools for managing bioinformatics data
https://github.com/fhcrc/seqmagick
bash shortcut:
% grep seqmagick ~/.bashrc
alias sc='seqmagick convert'
alias si='seqmagick info'
example usage:
% si seqs.fasta
name alignment min_len max_len avg_len num_seqs
seqs.fasta FALSE 5 120 12 71
% sc --head 2 seqs.fasta -
>seq1
GACGAACGCTGGCG
>seq2
CGTGCCTAATACATGCA
Command-line tools for bioinformatics analysis including sequence searching and clustering
https://github.com/torognes/vsearch
usage:
% vsearch --usearch_global query.fasta --db refs.fasta --alnout - --id 0 --maxaccepts 1 --strand both --quiet
vsearch v2.15.1_linux_x86_64, 503.6GB RAM, 128 cores
Query >seq1
%Id TLen Target
100% 1249 NZ_CNNQ02000124_1_1249
Query 332nt >seq1
Target 1249nt >NZ_CNNQ02000124
Qry 1 + CGTGGCTCAGGACGAACGCTGGCGGCGTGCCTAATACATGCAAGTAGAACGCTGAAGGAGGAGC 64
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tgt 17 + CCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAATACATGCAAGTAGAACGCTGAAGGAGGAGC 80
Qry 65 + TTGCTTCTCTGGATGAGTTGCGAACGGGTGAGTAACGCGTAGGTAACCTGCCTGGTAGCGGGGG 128
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tgt 81 + TTGCTTCTCTGGATGAGTTGCGAACGGGTGAGTAACGCGTAGGTAACCTGCCTGGTAGCGGGGG 144
Qry 129 + ATAACTATTGGAAACGATAGCTAATACCGCATAAGAGTGGATGTTGCATGACATTTGCTTAAAA 192
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tgt 145 + ATAACTATTGGAAACGATAGCTAATACCGCATAAGAGTGGATGTTGCATGACATTTGCTTAAAA 208
Qry 193 + GGTGCACTTGCATCACTACCAGATGGACCTGCGTTGTATTAGCTAGTTGGTGGGGTAACGGCTC 256
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tgt 209 + GGTGCACTTGCATCACTACCAGATGGACCTGCGTTGTATTAGCTAGTTGGTGGGGTAACGGCTC 272
Qry 257 + ACCAAGGCGACGATACATAGCCGACCTGAGAGGGTGATCGGCCACACTGGGACTGAGACACGGC 320
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tgt 273 + ACCAAGGCGACGATACATAGCCGACCTGAGAGGGTGATCGGCCACACTGGGACTGAGACACGGC 336
Qry 321 + CCAGACTCCTAC 332
||||||||||||
Tgt 337 + CCAGACTCCTAC 348
332 cols, 331 ids (99.7%), 0 gaps (0.0%)
Command-line tool for multiple alignment visualization
https://github.com/nhoffman/alnvu
% av seqs.afa
sequences 1 to 75 of 100
# 00000000000000000000000000000000000000000000000000
# 00000000000000000000000000000000000000000000000000
# 00000000111111111122222222223333333333444444444455
# 23456789012345678901234567890123456789012345678901
NC_003028pneumoniae556 TTAATGAGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAA
NC_010380pneumoniae630 TTAATGAGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAA
NC_011072pneumoniae750 TTAATGAGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAA
NZ_LN831051pneumoniaeT159 TTAATGAGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAA
NZ_KQ759755gordoniiT19 --AATGAGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAA
sequences 1 to 75 of 100
# 00000000000000000000000000000000000000000000000000
# 00000000000000000000000000000000000000000000000011
# 55555555666666666677777777778888888888999999999900
# 23456789012345678901234567890123456789012345678901
NC_003028pneumoniae556 TACATGCAAGTAGAACG.CTGA.AGGAGGAGCTTG...............
NC_010380pneumoniae630 TACATGCAAGTAGAACG.CTGA.AGGAGGAGCTTG...............
NC_011072pneumoniae750 TACATGCAAGTAGAACG.CTGA.AGGAGGAGCTTG...............
NZ_LN831051pneumoniaeT159 TACATGCAAGTAGAACG.CTGA.AGGAGGAGCTTG...............
NZ_KQ759755gordoniiT19 TACATGCAAGTAGAACG.Cac...aGtttAta---ccgtagctaccaacg
sequences 1 to 75 of 100
# 00000000000000000000000000000000000000000000000000
# 11111111111111111111111111111111111111111111111111
# 00000000111111111122222222223333333333444444444455
# 23456789012345678901234567890123456789012345678901
NC_003028pneumoniae556 .......CTtCTCT....................GGATGAGTTGCGAACG
NC_010380pneumoniae630 .......CTtCTCT....................GGATGAGTTGCGAACG
NC_011072pneumoniae750 .......CTtCTCT....................GGATGAGTTGCGAACG
NZ_LN831051pneumoniaeT159 .......CTtCTCTctacaccctacaccctacacGGATGAGTTGCGAACG
NZ_KQ759755gordoniiT19 ctacaccaTagaCT.....................Gtg-AGTTGCGAACG
Command-line tool for grouping pair-wise sequence alignments by taxonomy
https://github.com/crosenth/moose
bash shortcut:
% cat cls
#!/usr/bin/env bash
threads=100
dr blast blastn -num_threads $threads -db /path/to/nt -outfmt "10 qaccver saccver pident staxid" -query $1 | \
classify --columns "qaccver,saccver,pident,staxid" | \
./cl
example usage:
% cls query.fasta
|----------+---------------+----------------------------------------------+-----------+-------------+-------------+---------------+-------+----------+-----------|
| specimen | assignment_id | assignment | best_rank | max_percent | min_percent | min_threshold | reads | clusters | pct_reads |
|----------+---------------+----------------------------------------------+-----------+-------------+-------------+---------------+-------+----------+-----------|
| seq1 | 0 | Bacteria*;Streptococcus*;uncultured organism | species | 100.00 | 96.39 | 0.00 | 1 | 1 | 100.00 |
|----------+---------------+----------------------------------------------+-----------+-------------+-------------+---------------+-------+----------+-----------|
Command-line tool for comparing and identifying miss-annotated reference sequences
https://github.com/nhoffman/deenurp/blob/master/deenurp/subcommands/filter_outliers.py
visualization of data curated using filter_outliers:
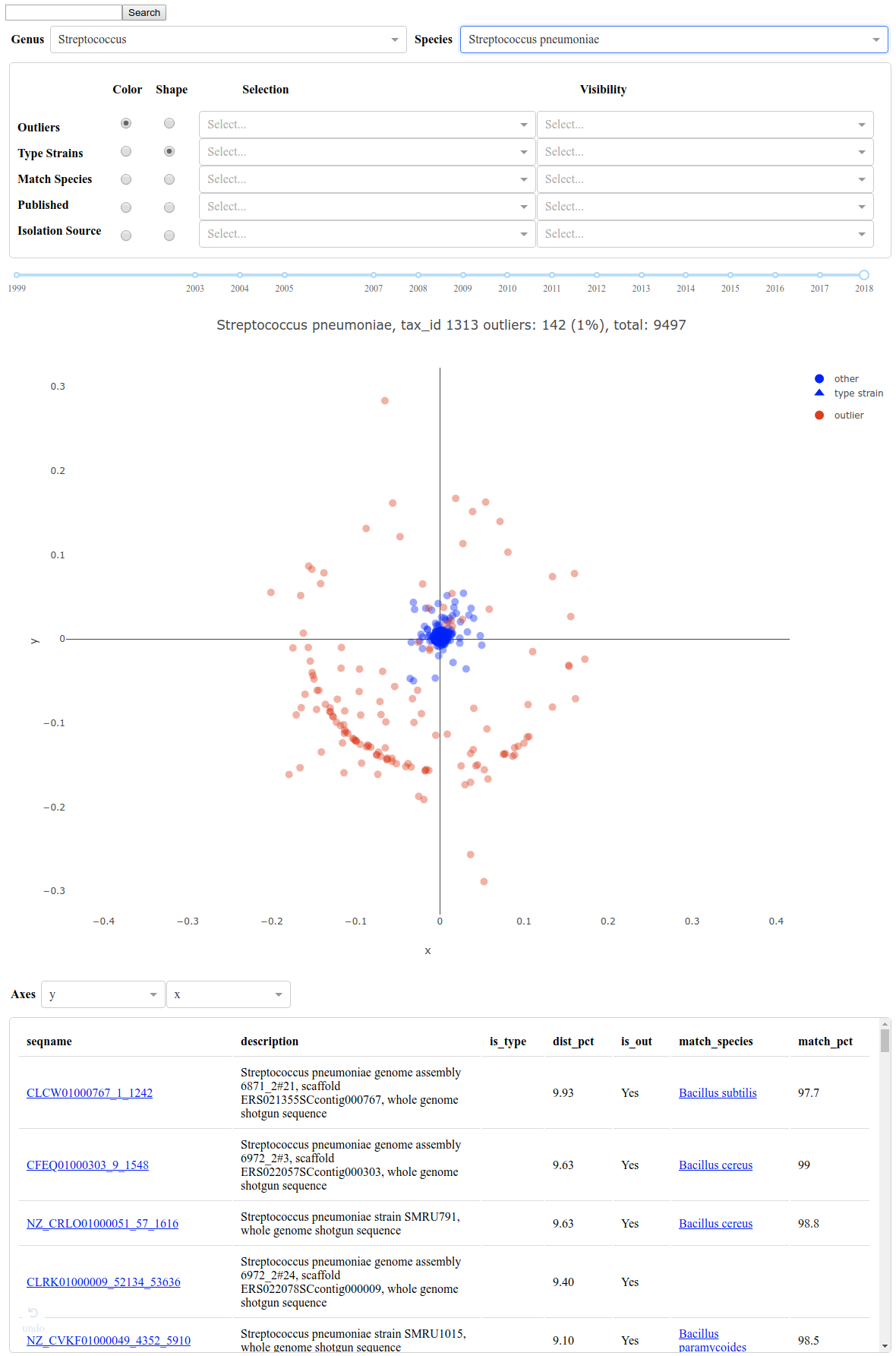
https://ya16sdb.labmed.uw.edu/